2.7.2. Реакции нуклеофильного замещения, протекающие по механизму SN2
За последние, годы существенно расширились наши знания о детальных механизмах реакций нуклеофильного замещения, широко распространенных в химии [117, 118]. Методами масс-спектрометрии высоких давлений [119], ионного циклотронного резонанса [120] и квантовой химии [121 - 123] показано, что в газовой фазе первоначально образуется устойчивый предреакционный комплекс (K1). Ему соответствует первый минимум на профиле потенциальной энергии. При дальнейшем движении вдоль координаты реакционная система преодолевает активационный барьер, после чего образуется второй комплекс (К2), который распадается на конечные продукты:
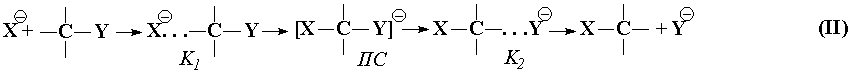
Таблица 2.2. Энтальпия (ΔН) и активационный барьер (Е#) реакции (III) в водном растворе (кДж/моль) [125]
|
X |
Н |
F |
Сl |
Н |
F |
Сl |
Н |
F |
Сl |
|
Y |
Н |
Н |
Н |
F |
F |
F |
Сl |
Сl |
Сl |
|
ΔН |
0 |
154 |
168 |
-154 |
0 |
17 |
168 |
-17 |
0 |
|
Е# |
224 |
280 |
220 |
126 |
66 |
144 |
50 |
127 |
65 |
Экспериментально эти реакции обычно изучаются в растворах. Влияние сольватации на профиль потенциальной энергии было установлено на основе результатов квантовохимических расчетов.
В работе [124] рассчитано сечение поверхности потенциальной энергии для реакции Сl- + Н3ССl → СlСН3 + Сl- неэмпирическим методом 3-21ГФ. Для моделирования сольватации использовано приближение супермолекулы (к реагентам добавлено две молекулы воды). Было показано, что сольватация существенно повышает активационный барьер реакции. Однако вычисленное значение энергии активации оказалось сильно заниженным по сравнению с экспериментом. Это связано, во-первых, с недостаточным количеством молекул воды, включенным в гидратационную оболочку реагентов, и, во-вторых, с использованием базиса 3-21 ГФ, расчеты в котором существенно переоценивают стабильность комплексов K1 и К2 и переходного состояния (ПС) реакции (II) в газовой фазе.
В работе [125] модифицированным методом ППДП/2 вычислен профиль потенциальной энергии для реакций [126],
X- + Н3СY → XСН3 + Y- (X,Y=H,F,Cl) (III)
для учета сольватации использована модель точечных зарядов. Полученные результаты приведены в табл. 2.2, из которой видно, что гидратация качественно изменяет профиль потенциальной энергии исследованных реакций и приводит к появлению активационных барьеров. Их высота, вычисленная квантовохимическим методом, близка к экспериментальным значениям.
В работе [127] профиль потенциальной энергии для реакции
Сl- + Н3ССl → СlСН3 + Сl- (IV)
ОН- + Н3СВr→ НОСН3 + Вr- (V)
рассчитан методом МПДП. Для учета гидратации использована модель точечных диполей. Полученные результаты приведены на рисунке 2.4 и в таблице 2.3. Видно, что гидратация существенно изменяет профиль потенциальной энергии исследованных реакций, однако траектория движения реагентов, которая проходит по дну долины на поверхности потенциальной энергии, при этом остается практически неизменной. Аналогичные результаты были получены в работах [128, 129], выполненных методом МПДП/Н с использованием приближения супермолекулы для расчета гидратации. Приблизительно такая же форма профиля потенциальной энергии для этих реакций получена и в работе [111], в которой расчеты проводились неэмпирическим методом в большом гауссовом базисе и методом Монте- Карло с атом-атомными потенциалами. Влияние сольватации на траекторию движения реагентов в последней работе не рассматривалось.
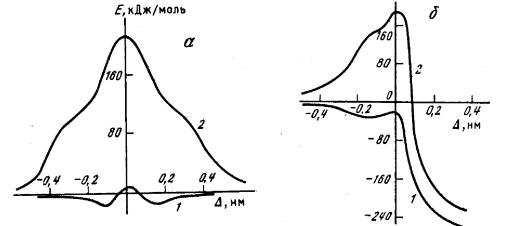
Рис. 2.4. Сечение ППЭ для реакций (IV) (a) и (V) (б)
1 – в газовой фазе; 2 – в водном растворе. Δ = R1 – R 2; R1 – расстояние между атомом углерода и уходящим атомом хлора (брома); R2 – расстояние между атомом углерода и атакующим атомом хлора (брома).
Таблица 2.3. Характеристики траектории движения реагентов в газовой фазе и в водном растворе
|
Газовая фаза |
Водный раствор |
Δ, нм |
||
|
Х...С |
C...Y |
Х...С |
C...Y |
|
|
Cl - + H3CCl |
ClCH3 + Cl - |
|
||
|
7,82 3,82 3,32 2,87 2,45 2,28 2,16 |
1,82 1,82 1,82 1,87 1,95 2,03 2,16 |
7,82 3,82 3,33 2,88 2,48 2,31 2,19 |
1,82 1,82 1,83 1,88 1,98 2,06 2,19 |
0,6 0,2 1,5 1,0 0,5 0,25 0 |
|
ОН -+СН3Вr |
Вr -+НОСН3 |
|
||
|
7,88 3,88 2,88 2,49 2,06 1,83 1,66 1,49 1,39 |
1,88 1,88 1,88 1,99 2,06 2,08 2,16 2,49 7,39 |
7,88 3,89 2,90 2,51 2,08 1,85 1,66 1,49 1,39 |
1,88 1,89 1,90 2,01 2,08 2,10 2,16 2,49 7,39 |
6 2 1 0,5 0 - 0,25 - 0,5 - 1 - 6 |
Таким образом, результаты, полученные разными методами, качественно совпадают. В связи с этим возникает вопрос, каким методом предпочтительнее проводить расчеты в прикладных работах: неэмпирическим методом и методом Монте-Карло или полуэмпирическими методами с использованием простых моделей? В первом случае результаты получаются более надежными, но расчеты очень трудно провести из-за недостаточного быстродействия современных ЭВМ, поэтому неэмпирические расчеты и расчеты методом Монте-Карло обычно проводят для существенно упрощенных модельных систем, которые далеки от экспериментально изучаемых реакций. Во втором случае расчеты удается проделать для более реалистичных моделей. Поэтому, с нашей точки зрения, для решения большинства прикладных задач следует пользоваться полуэмпирическими методами. Неэмпирические расчеты нужно проводить лишь для простейших модельных систем и тестировать по этим данным результаты полуэмпирических расчетов в тех случаях, когда необходимая экспериментальная информация отсутствует. Пример такого сочетания полуэмпирических и неэмпирических методов приведен ниже.