Активированного комплекса теория
АКТИВИРОВАННОГО КОМПЛЕКСА ТЕОРИЯ (теория переходного состояния, теория абс. скоростей р-ций), простейший и исторически первый вариант статистич. теории хим. р-ций. Разработана Э. Вигнером, М. Поляни, Г. Эйрингом, М. Эвансом в 30-х гг. 20 в. Позволяет приближенно рассчитывать скорость элементарных термич. хим. р-ций, исходя из электронного строения и св-в молекул реагентов. В основе теории лежит фундаментальное для химии понятие многомерной поверхности потенциальной энергии (ППЭ) р-ции. Для микроскопия, системы частиц (атомов, молекул), между к-рыми может происходить р-ция (в дальнейшем такую систему будем называть химической), ППЭ - ф-ция потенциальной энергии атомных ядер U от их внутр. координат, или степеней свободы. В системе из п ядер число внутр. степеней свободы N = 3n — 6 (или 3n — — 5, если все ядра расположены на одной прямой линии). Простейшая двухмерная (N = 2) ППЭ показана на рис. 1. Реагентам и продуктам р-ции на ней соответствуют области относительно небольшой потенциальной энергии (долины), разделенные областью повыш. энергии-потенциальным барьером. Кривая линия, проходящая по дну долин через барьер,-координата реакции. Часто используют одномерные схемы, изображающие сечение ППЭ, развернутое вдоль координаты р-ции (см. рис. 2). На этих схемах вершине потенциального барьера соответствует седло-вая точка, или точка перевала. Эти же понятия переносят на многомерные ППЭ с N > 2. Состояния реагентов и продуктов устойчивы, им соответствуют конфигурации (т.е. фиксированные значения координат ф), к-рые являются минимумами (или долинами) на многомерной ППЭ. Хим. р-ция рассматривается как переход из конфигурации реагентов в конфигурацию продуктов через конфигурацию седловой точки вдоль координаты р-ции. Конфигурации как минимумов, так и седловых точек-стационарные точки ППЭ, т.е. в них U/
U/ qi = 0.
qi = 0. 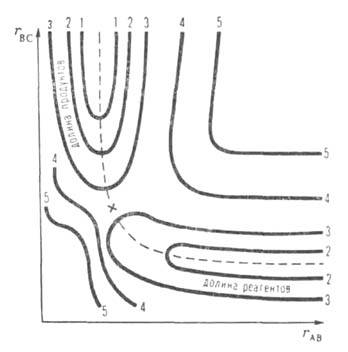
Рис. 1. Простейшая ППЭ для р-ции А + ВС -> АВ + С при расположении всех трех атомов А, В и С на одной прямой (угловые движения игнорируются). По координатным осям отложены межатомные расстояния rBC и rАВ- Кривые 1-5-уровни постоянной энергии (в условных единицах). Пунктиром обозначена координата р-ции, крестом-седловая точка.
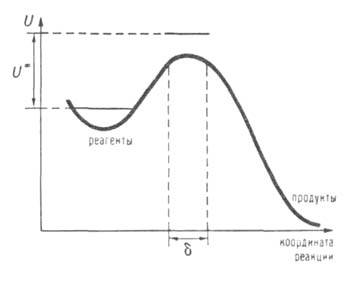
Рис. 2. Профиль ППЭ вдоль координаты р-ции. Вертикальные пунктирные линии ограничивают область, соответствующую активированному комплексу на координате р-ции (ее размер Горизонтальные сплошные линии-нулевые колебат. уровни энергии для реагентов и комплекса.
Горизонтальные сплошные линии-нулевые колебат. уровни энергии для реагентов и комплекса.
Активированного комплекса теория исходит из предположения о том, что скорость р-ции определяется св-вами нек-рой выделенной геом. конфигурации хим. системы. Обычно принимают, что эта конфигурация соответствует седловой точке на ППЭ; ее наз. активированным комплексом (АК), или переходным состоянием. На одномерной схеме (см. рис. 2) границы конфигурации АК определяются произвольно малой длиной 5 вблизи вершины барьера.
Уравнение для скорости реакции. В седловой точке ППЭ с координатами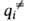 (i = 1, 2, ..., N)все первые производные ф-ции U(qi)равны нулю, так что разложение ф-ции по отклонениям (qi —
(i = 1, 2, ..., N)все первые производные ф-ции U(qi)равны нулю, так что разложение ф-ции по отклонениям (qi —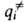 ) начинается с квадратичных членов. Это означает, что для АК можно определить N нормальных колебаний (мод), как и для обычных устойчивых молекул, а также ввести вращат. и постулат. степени свободы, характеризующие движение АК как целого. Однако в отличие от устойчивой молекулы, к-рой соответствует на ППЭ не сед-ловая точка, а минимум, для АК одна из нормальных мод, а именно движение вдоль координаты р-ции, приводит к его распаду; частота этой моды-чисто мнимая величина. Неустойчивая конфигурация АК характеризуется временем жизни:
) начинается с квадратичных членов. Это означает, что для АК можно определить N нормальных колебаний (мод), как и для обычных устойчивых молекул, а также ввести вращат. и постулат. степени свободы, характеризующие движение АК как целого. Однако в отличие от устойчивой молекулы, к-рой соответствует на ППЭ не сед-ловая точка, а минимум, для АК одна из нормальных мод, а именно движение вдоль координаты р-ции, приводит к его распаду; частота этой моды-чисто мнимая величина. Неустойчивая конфигурация АК характеризуется временем жизни: 
где k и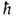 -постоянные Больцмана и Планка соотв., Т-абс. т-ра. При обычных для хим. р-ций т-рах
-постоянные Больцмана и Планка соотв., Т-абс. т-ра. При обычных для хим. р-ций т-рах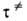 ~10-13с.
~10-13с.
Активированного комплекса теория постулирует термодинамич. равновесие между реагентами и АК, характеризуемое константой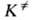 . На этом основании константа скорости хим. р-ции к выражается ур-ниями:
. На этом основании константа скорости хим. р-ции к выражается ур-ниями:
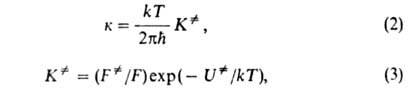
где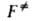 и F-отнесенные к единице объема статистич. суммы АК и реагентов соотв.,
и F-отнесенные к единице объема статистич. суммы АК и реагентов соотв.,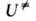 -изменение потенциальной энергии системы при переходе от реагентов к АК. Величина
-изменение потенциальной энергии системы при переходе от реагентов к АК. Величина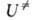 складывается из высоты потенциального барьера и разности нулевых колебат. энергий АК и реагентов (см. рис. 2). В ф-ле (3) она приведена в расчете на 1 АК; обычно же ее относят к NA = 6,02*1023 АК; тогда в показателе экспоненты k заменяют газовой постоянной R. При вычислении статистич. суммы
складывается из высоты потенциального барьера и разности нулевых колебат. энергий АК и реагентов (см. рис. 2). В ф-ле (3) она приведена в расчете на 1 АК; обычно же ее относят к NA = 6,02*1023 АК; тогда в показателе экспоненты k заменяют газовой постоянной R. При вычислении статистич. суммы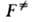 учитывают все степени свободы АК, кроме движения по координате р-ции, а именно постулат. и вращат. движения АК как целого и (N — 1) остальных колебаний, наз. поперечными.
учитывают все степени свободы АК, кроме движения по координате р-ции, а именно постулат. и вращат. движения АК как целого и (N — 1) остальных колебаний, наз. поперечными.
В ранних формулировках активированного комплекса теории гипотеза о равновесии между реагентами и АК трактовалась буквально и к при равнивалась произведению частоты распада АК 1/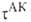 на константу равновесия К :
на константу равновесия К :

Величины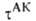 и КАК вычисляются обычными методами статистич. механики; обе они пропорциональны длине
и КАК вычисляются обычными методами статистич. механики; обе они пропорциональны длине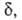 определяющей границы конфигурации АК на координате р-ции. Поскольку их отношение (4) не зависит от
определяющей границы конфигурации АК на координате р-ции. Поскольку их отношение (4) не зависит от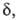 эти границы условны, ур-ния (2) и (3) получаются из (4) при произвольном выборе значения
эти границы условны, ур-ния (2) и (3) получаются из (4) при произвольном выборе значения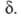 Выражение К через статистич. суммы аналогично ф-ле (3), но вместо
Выражение К через статистич. суммы аналогично ф-ле (3), но вместо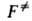 используют полную статистич. сумму АК F = f
используют полную статистич. сумму АК F = f , где f -элементарная статистич. сумма для движения системы вдоль координаты р-ции; именно она пропорциональна
, где f -элементарная статистич. сумма для движения системы вдоль координаты р-ции; именно она пропорциональна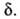 Можно ввести спец. определение АК, выбрав
Можно ввести спец. определение АК, выбрав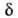 т. обр., чтобы для
т. обр., чтобы для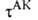 и К получились в точности выражения (1) и (3), т.е. положив
и К получились в точности выражения (1) и (3), т.е. положив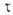 АК =
АК =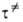 и КАК =
и КАК =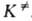 . При этом f= 1 и FAK =
. При этом f= 1 и FAK =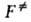 . Тогда термодинамич. интерпретация ур-ния (2) становится особенно наглядной; соответствующее значение
. Тогда термодинамич. интерпретация ур-ния (2) становится особенно наглядной; соответствующее значение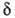 составляет ок. 1 нм.
составляет ок. 1 нм.
Рассматривая в (3)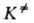 как константу равновесия, можно представить (2) в термодинамич. форме:
как константу равновесия, можно представить (2) в термодинамич. форме: 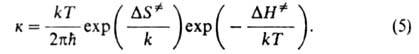
Величины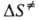 и
и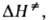 наз. соотв. энтропией и энтальпией активации, представляют собой изменения энтропии и энтальпии системы при переходе от реагентов к АК. Как правило, осн. вклад в
наз. соотв. энтропией и энтальпией активации, представляют собой изменения энтропии и энтальпии системы при переходе от реагентов к АК. Как правило, осн. вклад в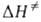 дает
дает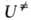 , а
, а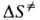 определяется в осн. статистич. суммами:
определяется в осн. статистич. суммами: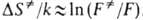 ; исключением м.б. р-ции в полярных р-рителях. Ур-ния (2) и (3) применяют к газофазным р-циям, а (5)-для расчета скоростей р-ций в р-рах, когда вычисления статистич. сумм затруднительны. Соотв. предполагается, что в первом случае р-ция протекает при постоянном объеме, во втором-при постоянном давлении.
; исключением м.б. р-ции в полярных р-рителях. Ур-ния (2) и (3) применяют к газофазным р-циям, а (5)-для расчета скоростей р-ций в р-рах, когда вычисления статистич. сумм затруднительны. Соотв. предполагается, что в первом случае р-ция протекает при постоянном объеме, во втором-при постоянном давлении.
Совр. вывод ур-ния (2), химически менее наглядный, основан на столкновений теории. Скорость р-ции отождествляется со скоростью перехода реагирующих хим. систем через (N — 1 )-мерную пов-сть в пространстве конфигураций, разделяющую области реагентов и продуктов. В теории столкновений эта скорость наз. потоком через критич. пов-сть. Ур-ние в форме (2) получается, если провести критич. пов-сть через седловую точку ортогонально координате р-ции и принять, что на критич. пов-сти энергетич. распределение реагентов равновесно. Соответствующая область пространства координат и импульсов (фазового пространства) характеризуется той же статистич. суммой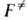 . Это позволяет рассматривать критич. пов-сть как множество конфигураций АК. Т. обр., АК сразу определяется как объект с (N — 1) внутр. степенями свободы и не нужно вводить его протяженность
. Это позволяет рассматривать критич. пов-сть как множество конфигураций АК. Т. обр., АК сразу определяется как объект с (N — 1) внутр. степенями свободы и не нужно вводить его протяженность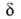 вдоль координаты р-ции.
вдоль координаты р-ции.
Применение теории. Согласно теории, механизм р-ции вполне определен конфигурациями реагентов и продуктов (минимумы, или долины, на ППЭ) и соответствующих АК (седловые точки). Теоретич. расчет этих конфигураций методами квантовой химии дал бы исчерпывающую информацию о направлениях и скоростях хим. р-ций. Такие расчеты интенсивно развиваются; для простых хим. систем, содержащих 10-15 атомов, к-рые принадлежат к элементам первых двух периодов таблицы Менделеева, они практически реализуемы и достаточно надежны. Последоват. расчет абс. скорости р-ции по ур-нию (2) заключается в определении геом. конфигураций реагентов и АК (на этом этапе также определяется высота потенциального барьера) и вычислении для этих конфигураций моментов инерции и колебат. частот, к-рые необходимы для расчета статистич. сумм и окончат. определения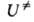 . В применении к сложным р-циям, представляющим практич. интерес, полная и надежная реализация такой программы трудоемка и зачастую неосуществима. Поэтому молекулярные постоянные, необходимые для вычислений по ур-ниям (2) и (3), часто находят эмпирич. методами. Для устойчивых конфигураций реагентов моменты инерции и колебат. частоты обычно известны из спектроскопич. данных, однако для АК эксперим. определение их невозможно ввиду малого впемени его жизни. Если последоват. квантовохим. расчет
. В применении к сложным р-циям, представляющим практич. интерес, полная и надежная реализация такой программы трудоемка и зачастую неосуществима. Поэтому молекулярные постоянные, необходимые для вычислений по ур-ниям (2) и (3), часто находят эмпирич. методами. Для устойчивых конфигураций реагентов моменты инерции и колебат. частоты обычно известны из спектроскопич. данных, однако для АК эксперим. определение их невозможно ввиду малого впемени его жизни. Если последоват. квантовохим. расчет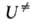 и
и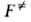 недоступен, для оценки этих величин применяют интерполяционные расчетные схемы.
недоступен, для оценки этих величин применяют интерполяционные расчетные схемы.
Активированного комплекса теория - основа качеств. представлений о реакц. способности в-в. Ур-ния (2), (3) и (5) имеют ту же форму, что и ур-ние Аррениуса, к-рое эмпирически описывает температурную зависимость кинетич. констант разл. хим. процессов. Величину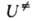 во мн. случаях достаточно отождествить с наблюдаемой энергией активации, пренебрегая слабой (по сравнению с экспоненциальной) температурной зависимостью статистич. сумм и множителя k Т/
во мн. случаях достаточно отождествить с наблюдаемой энергией активации, пренебрегая слабой (по сравнению с экспоненциальной) температурной зависимостью статистич. сумм и множителя k Т/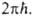 Тогда пред-экспоненциальный множитель в ур-ниях (2) и (3) можно отождествить с аррениусовским. Его значение слабо зависит от деталей строения АК и оценка по порядку величины не составляет труда. Оказывается, что в реакц. сериях с одинаковым реакц. центром предэкспоненциальный множитель примерно постоянен, т.е. ряды активности определяются значениями энергии активации. Наконец, если пренебречь вкладом нулевых колебат. энергий в
Тогда пред-экспоненциальный множитель в ур-ниях (2) и (3) можно отождествить с аррениусовским. Его значение слабо зависит от деталей строения АК и оценка по порядку величины не составляет труда. Оказывается, что в реакц. сериях с одинаковым реакц. центром предэкспоненциальный множитель примерно постоянен, т.е. ряды активности определяются значениями энергии активации. Наконец, если пренебречь вкладом нулевых колебат. энергий в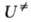 , высота потенциального барьера р-ции становится единственной фундам. характеристикой ее скорости. Для теоретич. оценки относит. изменений высоты потенциального барьера в реакц. сериях разработаны простые методы (см. Реакционная способность). Такой подход к оценке относит. скоростей применяют для любого физ.-хим. процесса, если высота потенциального барьера, разделяющего исходное и конечное состояния, достаточно высока по сравнению с kT; он не требует громоздких вычислений и широко распространен. Именно этим определяется плодотворность и универсальность концепции АК в теоретич. химии.
, высота потенциального барьера р-ции становится единственной фундам. характеристикой ее скорости. Для теоретич. оценки относит. изменений высоты потенциального барьера в реакц. сериях разработаны простые методы (см. Реакционная способность). Такой подход к оценке относит. скоростей применяют для любого физ.-хим. процесса, если высота потенциального барьера, разделяющего исходное и конечное состояния, достаточно высока по сравнению с kT; он не требует громоздких вычислений и широко распространен. Именно этим определяется плодотворность и универсальность концепции АК в теоретич. химии.
Ограниченность теории и попытки ее совершенствования. Активированного комплекса теория основана на двух предположениях. Первое-гипотеза о термодинамич. равновесии между реагентами и АК. Согласно второму, скорость р-ции отождествляется со скоростью распада АК. Оба предположения нельзя строго обосновать. Это обнаруживается, если рассматривать движение хим. системы вдоль координаты р-ции на всем пути от реагентов к продуктам, а не только вблизи вершины потенциального барьера. Координату р-ции лишь в редких случаях правильно считать прямой линией, как на рис. 2. Обычно же она-кривая в многомерном пространстве внутр. переменных и является сложной комбинацией элементарных движений, к-рая неодинакова на разл. своих участках. Напр., на рис. 1 координата р-ции-это непрерывно изменяющаяся комбинация двух валентных колебаний.
Равновесное распределение энергии в реагентах для термич. р-ций обеспечено практически всегда; оно нарушается только в чрезвычайно быстрых процессах. Проблема в том, сохранится ли оно в АК. Из-за криволинейности координату р-ции нельзя считать независимой степенью свободы. Ее взаимод. с другими, поперечными движениями приводит к обмену энергией между ними. В результате, во-первых, может нарушиться первоначально равновесное распределение энергии по поперечным степеням свободы и, во-вторых, система может вернуться в область реагентов даже после того, как она уже прошла через конфигурацию АК в направлении продуктов. Наконец, необходимо иметь в виду, что, согласно ур-ниям (2), (3) и (5), хим. р-ция рассматривается как классич. переход; игнорируются квантовые особенности, напр. электронно-неадиабатич. процессы и туннельный эффект. В ранних формулировках теории в ур-ния (2), (3) и (5) добавляли т. наз. трансмиссионный множитель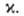 Предполагалось, что в нем собрано влияние перечисленных выше факторов, не учтенных при выводе этих ур-ний. Т. обр., определение х выходит за рамки активированного комплекса теории; более того, для р-ций, в к-рых х значительно отличается от единицы, теория теряет смысл. Однако для сложных р-ций предположение
Предполагалось, что в нем собрано влияние перечисленных выше факторов, не учтенных при выводе этих ур-ний. Т. обр., определение х выходит за рамки активированного комплекса теории; более того, для р-ций, в к-рых х значительно отличается от единицы, теория теряет смысл. Однако для сложных р-ций предположение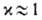 не противоречит экспе-рим. данным, и именно этим объясняется популярность активированного комплекса теории.
не противоречит экспе-рим. данным, и именно этим объясняется популярность активированного комплекса теории.
Последоват. неформальное рассмотрение всех указанных эффектов возможно лишь в рамках динамич. расчета (см. Динамика элементарного акта). Предпринимались попытки учесть их по отдельности. Напр., был предложен метод си-стематич. уточнения конфигурации АК, поскольку выбор в кач-ве таковой именно седловой точки основан на интуитивных представлениях и, вообще говоря, не обязателен. Могут существовать и др. конфигурации, для к-рых погрешность вычислений по ф-лам (2) и (3), обусловленная возвращением системы в область реагентов после прохождения этих конфигураций, меньше, чем для конфигурации седловой точки. Используя формулировку активированного комплекса теории в терминах теории столкновений (см. выше), можно утверждать, что обратному потоку (от продуктов к реагентам) через критич. пов-сть соответствует порождающая его и равная ему часть полного прямого потока (от реагентов к продуктам). Чем меньше эта часть, тем точнее вычисление скорости р-ции по активированного комплекса теории. Эти соображения легли в основу т. наз. вариационного определения АК, согласно к-рому критической считается пов-сть, минимизирующая прямой поток. Для нее скорость р-ции, вычисляемая по ур-ниям (2) и (3), минимальна. Как правило, нулевые энергии поперечных колебаний изменяются вдоль координаты р-ции. Это еще одна причина смещения конфигурации АК из седловой точки ППЭ; она также учитывается вариационной теорией.
Значит. внимание уделялось разработке методов определения вероятностей квантового туннелирования в хим. р-циях. Наконец, стали возможны оценки трансмиссионного множителя в рамках модельных динамич. вычислений. При этом предполагается, что с постулат. движением системы вдоль координаты р-ции взаимодействуют не все, а лишь нек-рые из поперечных степеней свободы. Они и учитываются в квантовом динамич. расчете; остальные степени свободы обрабатываются в рамках равновесной теории. При таких вычислениях автоматически определяются также и поправки на квантовое туннелирование.
Упомянутые усовершенствованные методы расчета абс. скоростей хим. р-ций требуют серьезных вычислит. усилий и лишены универсальности активированного комплекса теории.
Литература
Глесстон С, Лейдлер К., Эйринг Г., Теория абсолютных скоростей реакций, пер. с англ., М., 1948; Лейдлер К., Кинетика органических реакций, пер. с англ., М., 1966: Термические бимолекулярные реакции в газах, М., 1976. М. В. Базилевский.