Неравновесная химическая кинетика
НЕРАВНОВЕСНАЯ ХИМИЧЕСКАЯ КИНЕТИКА, изучает кинетич. закономерности хим. р-ций при сильном нарушении термодинамич. равновесия в реагирующей системе или физ.-хим. среде, в к-рой они протекают. Любая хим. р-ция нарушает термодинамич. равновесие в системе, но во мн. случаях это нарушение мало, и если нет внеш. источников возмущения состояния системы, то при кинетич. расчетах неравновесностью либо пренебрегают, либо учитывают как второстепенный фактор, вводя малые поправки к константам скорости р-ций. В таких случаях говорят о р а в н о в е с н о й к и н е т и к е (условно, поскольку хим. состав системы должен быть неравновесным, иначе скорости всех р-ций были бы равны нулю). Константы скорости в равновесной кинетике выражаются в виде ф-ций от термодинамич. параметров среды, напр. т-ры и давления.
В условиях термодинамич. равновесия относит. заселенность i-го энергетич. уровня Ni /N (N-полное число молекул, Ni—число молекул на энергетич. уровне Ei)зависит от т-ры Т окружающей среды и описывается ф-л о й Б о л ь ц-м а н а:
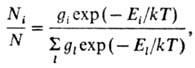
где k-постоянная Больцмана, gi, gl-числа возможных квантовых состояний молекулы на уровнях с энергиями Еi и Еl соотв. (суммирование проводится по индексу l). Если оператор энергии (гамильтониан) системы допускает разделение переменных, то энергию Еi можно выразить в виде суммы энергий независимых или слабо связанных подсистем. В газах Еi можно представить в виде суммы энергий поступат. и вращат. движений молекул, внутримол. колебаний и электронного возбуждения, причем каждый вид энергии описывается своей ф-цией распределения типа ф-лы Больцмана.
При отклонении от равновесия все или нек-рые из распределений частиц по энергиям типа больцмановского не реализуются. Это может приводить к качеств. и количеств. изменениям кинетики р-ций. Не существует признаков, позволяющих разделить хим. кинетику на неравновесную и равновесную. Строго судить об этом нельзя ни по величине относит. отклонения заселенности конкретных энергетич. уровней от равновесной заселенности Ni /N, ни по числу таких уровней, ни по отличию константы скорости р-ции от ее равновесного значения и т.п. В общем случае можно говорить только об условных границах в зависимости от требуемой точности решения конкретной кинетич. задачи. Однако по мере удаления от таких "размытых" границ признаки неравновесных хим. р-ций становятся все более определенными и м. б. установлены на основе общих качеств. сопоставлений характерных времен релаксационных процессов в газах и конденсир. средах (т. е. по иерархии времен релаксации). Система, выведенная из состояния тер-модинамич. равновесия, возвращается к нему (релаксирует) в результате обмена энергией при столкновениях частиц неравномерно, с перераспределением по типам движений (степеням свободы молекулы). В газах равновесие м. б. достигнуто быстрее всего для поступат. движения частиц, имеющих одинаковые (или близкие по величине) массы. Колебат. движение, как правило, не обменивается энергией с поступат. движением в процессе столкновения. Молекула может претерпеть значит.число столкновений, прежде чем она приобретет или потеряет квант колебат. энергии. Обычно в системе сначала устанавливается общее равновесие поступат. и вращат. движений. Колебат. релаксация, ведущая к равновесию колебаний молекул с их поступат. и вращат. движениями, требует значительно большего времени.
Если в газовой смеси имеются частицы, на порядки величин различающиеся по массам, время установления поступат. равновесия для смеси в целом гораздо больше, чем для отдельных компонент осн. состава (но не малых примесей). В ионизир. газе вследствие огромного различия масс электронов и атомов задолго до завершения поступат. релаксации устанавливаются равновесия отдельно в подсистемах "тяжелых" частиц (атомов и ионов) и электронов с двумя в общем случае разными т-рами, соотв. Тaпост и Тэлпост. В процессе колебат. релаксации до его завершения в зависимости от состава смеси и типа колебаний могут устанавливаться равновесия по отдельным группам колебат. степеней свободы (колебат. подсистемам), каждое со своей т-рой Ткол, и между такими группами.
Таким же соотношением (иерархией) времен релаксации характеризуются мол. жидкости и мол. кристаллы, с тем, однако,отличием, что в жидкостях поступат. и вращат. движения молекул составляют обычно единое целое (энергии этих видов движения можно разделить лишь в очень грубом приближении). В твердых телах все движения атомов и молекул колебательные.
Равновесие по хим. составу, к к-рому приводят хим. р-ции, как правило, достигается за времена, значительно большие по сравнению со временем колебат. релаксации. Однако при достаточно высоких т-рах константы скорости р-ций сильно возрастают как по абс. величине (см. Аррениуса уравнение), так и в сравнении с временами др. релаксационных процессов, и в системе создаются условия, при к-рых возмущения, вызываемые хим. р-цией, релаксировать не успевают. Это относится в особенности к релаксации тех энергетич. состояний, от заселенности к-рых зависит скорость р-ции. В результате скорость р-ции становится зависящей от времен колебат. релаксации, а иногда, в предельных случаях сильной неравновесности, и от времен вращат. и поступат. релаксаций. Иными словами, устанавливается отрицат. обратная связь между скоростью р-ции и теми возмущениями, к-рые она вызывает, что можно рассматривать как одно из проявлений Ле Шателье - Брауна принципа. Зависимость скорости р-ции от Тпост становится при этом более слабой. Так, в сильных ударных волнах константа диссоциации при высоких Тпост(в условиях kTnocт>= D/17, где D- энергия диссоциации) обычно выражается соотношением
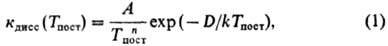
где показатель степени п принимает значения от 1 до 4, A-эмпирич. постоянная.
Количеств. соотношения неравновесной химической кинетики, как правило, значительно сложнее, чем равновесной. Кроме кинетич. ур-ний для концентраций реагирующих в-в и продуктов приходится иметь дело и с ур-ниями, выражающими временные зависимости для заселенностей возбужденных состояний частиц. Такие системы ур-ний, в принципе, можно решать на ЭВМ, если имеются данные о константах скорости элементарных процессов мол. переноса энергии - колебательно-вращательного, колебательно-поступательного (колебательно-трансляционного) и вращательно-трансляционного. В сильно неравновесных условиях решение задач неравновесной химической кинетики обеспечивается не столько возможностью решения полной системы ур-ний для заселенностей всех энергетич. состояний частиц, сколько правильным выделением "узкого места" в совокупности элементарных актов, из к-рых слагается хим. превращение. Для этого нужно определить наиб. быстрые параллельные и наиб. медленные последовательные переходы и вычислить (или измерить) их константы скорости - величины, обратные временам жизни молекул в соответствующих возбужденных состояниях.
Вычисление констант скорости хим. р-ций упрощается, если в неравновесной в целом системе можно выделить равновесные подсистемы. Константы скорости р-ций в таких случаях выражаются как ф-ции т-р подсистем. Напр., константа скорости диссоциации кABдисс двухатомного газа АВ при высоких Tпост приближенно представляется в виде ф-лы типа (1), но в экспоненциальный множитель вместо Тпост входит Ткол, а предэкспоненциальный множитель слабо зависит от т-ры. Т. к. движение атомов в молекуле носит в осн. колебат. характер, а кинетика многих хим. превращений связана именно с внутримол. перемещениями атомов, т-ра Ткол- важнейшая кинетич. и энергетич. характеристика состояния газа в условиях, описываемых неравновесной химической кинетикой. Для молекул, состоящих из неск. атомов, константа скорости мономол. распада м. б. при низких давлениях экспоненц. ф-цией Тпост и Tкол, общей для всех колебаний (иногда отдельно рассматриваются Ткол для низко- и высокочастотных колебаний).
В случае р-ций с участием электронов (ионизация А + е А+ + 2е, диссоциативная рекомбинация АВ+ + е
А+ + 2е, диссоциативная рекомбинация АВ+ + е А + В и др.) обычно сравнительно быстро устанавливаются электронное равновесие, характеризующееся т-рой Тэлпост, и поступат. равновесие, характеризующееся т-рой Тапост. Константа скорости ионизации атома А с точностью до слабо меняющегося предэкспоненциального множителя пропорциональна exp(-I/kTэлпост), где I-потенциал ионизации. Связь между Ткол и Тпост (или Тэлпост и Тапост) определяется ур-нием баланса энергии каждой из подсистем, в к-ром учитывается их взаимод. и вклад подсистемы в энергетику р-ций. Электроны обмениваются энергией с колебаниями эффективнее, чем с поступат. и вращат. движениями, поэтому до установления полного равновесия в системе может наступить равновесие между электронной и колебат. подсистемами, выражающееся в равенстве Ткол = Тэлпост. Определяя Тэлпост, напр. по данным о свечении газа, можно косвенно оценить Ткол.
А + В и др.) обычно сравнительно быстро устанавливаются электронное равновесие, характеризующееся т-рой Тэлпост, и поступат. равновесие, характеризующееся т-рой Тапост. Константа скорости ионизации атома А с точностью до слабо меняющегося предэкспоненциального множителя пропорциональна exp(-I/kTэлпост), где I-потенциал ионизации. Связь между Ткол и Тпост (или Тэлпост и Тапост) определяется ур-нием баланса энергии каждой из подсистем, в к-ром учитывается их взаимод. и вклад подсистемы в энергетику р-ций. Электроны обмениваются энергией с колебаниями эффективнее, чем с поступат. и вращат. движениями, поэтому до установления полного равновесия в системе может наступить равновесие между электронной и колебат. подсистемами, выражающееся в равенстве Ткол = Тэлпост. Определяя Тэлпост, напр. по данным о свечении газа, можно косвенно оценить Ткол.
Равновесное распределение колебат. энергии в двухатомном газе (молекулы АВ) при Ткол 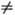 Тпост осуществляется путем быстрого (почти резонансного) обмена колебат. квантами по схеме: АВn + АВm
Тпост осуществляется путем быстрого (почти резонансного) обмена колебат. квантами по схеме: АВn + АВm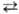 ABnb1 + АВтb1 (индексами обозначены номера колебат. уровней). При таком обмене сохраняется общее число колебат. квантов, поэтому равновесие, к к-рому обмен приводит, характеризуется не только т-рой Тпост, но и хим. потенциалом m
ABnb1 + АВтb1 (индексами обозначены номера колебат. уровней). При таком обмене сохраняется общее число колебат. квантов, поэтому равновесие, к к-рому обмен приводит, характеризуется не только т-рой Тпост, но и хим. потенциалом m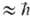 w0 (1 - Тпост/Ткол), где
w0 (1 - Тпост/Ткол), где 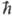 w0 = E1 - E0 (w0- основная частота колебаний). Для колебат. уровней с энергиями Ei, меньшими нек-рого значения Ei* (номер уровня i
w0 = E1 - E0 (w0- основная частота колебаний). Для колебат. уровней с энергиями Ei, меньшими нек-рого значения Ei* (номер уровня i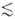 i*), обмен энергии внутри колебат. подсистемы происходит быстрее, чем между колебаниями и поступат. движением молекул. Заселенности Xi таких уровней определяются выражением
i*), обмен энергии внутри колебат. подсистемы происходит быстрее, чем между колебаниями и поступат. движением молекул. Заселенности Xi таких уровней определяются выражением
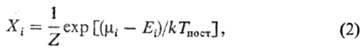
где 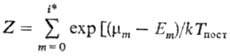 .
.
Константа скорости диссоциации при этом выражается в виде
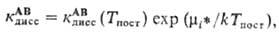
гдe кАВдисс(Тпост)-значение кАВдисс при Ткол = Тпост. Распределение (2) переходит в ф-лу Больцмана при Ткол = Тпост.
В бинарной газовой смеси двухатомных молекул АВ и CD с близкими частотами колебаний происходит быстрый обмен колебат. квантами при столкновениях как одинаковых, так и разных молекул. При этом устанавливается след. соотношение между колебат. т-рами ТколАВ и ТколCD :
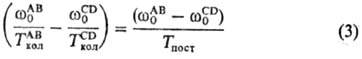
Благодаря соотношениям (2) и (3) система ур-ний неравновесной химической кинетики существенно упрощается, т. к. заселенности Хi не являются независимыми переменными и выражаются через небольшое число параметров - Tпост и m (или Тпост и Ткол).
Выделившаяся при экзотермич. р-ции энергия во мн. случаях распределена по квантовым состояниям продуктов не статистически. Но даже в случае статистич. распределения оно неравновесно относительно окружающей среды. Если в послед. р-ции вступают гл. обр. те промежут. частицы, к-рые не успели релаксировать (дезактивироваться), то р-ция в целом описывается в рамках неравновесной химической кинетики. В частности, к объектам неравновесной химической кинетики относятся цепные р-ции, если активными центрами, ведущими р-цию, являются неравновесно возбужденные частицы. Т. наз. энергетич. разветвление цепей было экспериментально обнаружено в смесях Н2 с F2 и СН3I с F2. В смеси Н2 с F2 разветвление обусловлено генерированием колебательно возбужденной молекулы HF* по схеме: F + Н2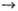 HF* + Н с почти резонансной быстрой передачей этого возбуждения на молекулу Н2 и послед. прямой р-цией:
HF* + Н с почти резонансной быстрой передачей этого возбуждения на молекулу Н2 и послед. прямой р-цией:
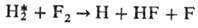
По законам неравновесной химической кинетики происходят нек-рые каталитич. процессы, если активными центрами на каталитич. пов-сти служат адсорбир. частицы в колебательно- или электронно-возбужденных состояниях.
Распределение колебат. энергии реагента и константа скорости мономол. р-ции (распада, изомеризации) взаимно связаны друг с другом в переходной области давлений и в пределе низких давлений. Механизм неравновесной химической кинетики лежит в основе хим. превращений конденсир. систем в ударных волнах, нек-рых механохим. процессов (см. Механохимия). Особое место занимает неравновесная химическая кинетика р-ций, приводящих к образованию колебаний концентраций промежут. в-в и (или) продуктов (см. Колебательные реакции).
Неравновесность в физ.-хим. среде возникает практически во всех случаях, когда на скорость и характерные особенности хим. р-ций (напр., селективность) воздействуют физ. поля. Это м. б. электрич. поле (дуговой разряд, высокочастотное и СВЧ перем. поле), электромагн. излучение ИК, УФ, рентгеновского диапазонов частот, ионизирующее излучение (g-кванты, др. жесткая радиация). Электромагн. излучение взаимод. с электронной подсистемой, приводя к электронному возбуждению атомов и молекул, ионизации частиц, увеличению энергии своб. электронов (т-ра Тэлпост) и, как следствие, к увеличению энергии мол. колебаний (т-ры Ткол). ИК излучение может и непосредственно возбуждать оптически разрешенные (излучательныe) колебат. переходы.
Если источник возбуждения взаимод. не со всеми, а лишь с нек-рыми подсистемами, то при их относительно медленной релаксации имеется возможность направленного (селективного) возбуждения таких подсистем. Высокая монохроматичность лазерного ИК излучения позволяет возбуждать отдельные типы колебаний в молекуле, находящиеся в резонансе с излучением. Поскольку вероятность того, что колебат. энергия выделится в результате спонтанного ИК излучения, обычно очень мала, лазерное колебат. возбуждение способно приводить к очень высокой колебат. т-ре.
Ангармонизм колебаний и перераспределение энергии между разл. степенями свободы при соударениях молекул приводят к ограничению направленности действия источника возбуждения системы. Для достижения наиб. выхода продукта при минимуме затрат энергии нужно, как правило, возбуждать не одну, а неск. определенных колебат. степеней свободы, причем не обязательно оптически разрешенных. Это позволяет управлять хим. р-циями: их скоростью, составом продукта и др. Подобные задачи решаются, в частности, в плазмохимии, фотохимии, радиационной химии, лазерной химии. Первичные продукты внеш. воздействия -сильно неравновесные по хим. составу и степени возбуждения частицы - могут, взаимодействуя, приводить к образованию больших концентраций др. возбужденных частиц, в т. ч. с инверсной заселенностью, что является необходимым условием для генерирования лазерного излучения (см. Лазеры химические).
Подходы неравновесной химической кинетики плодотворны для описания мн. прир. процессов. Так, на больших высотах в атмосфере в дневное время суток под действием солнечной радиации происходит эндотермич. диссоциация О2 и N2, а в ночное время преобладают обратные процессы с выделением аккумулированной солнечной энергии. Ниже (на высотах 25-35 км) формируется озонный слой. Во всех процессах, от к-рых зависит состав верх. слоев атмосферы, тепловой режим Земли, климат и погода, спектр. состав излучения у земной поверхности и т. п., важную роль играют возбужденные состояния молекул и атомов, их повышенная реакц. способность. Во многом благодаря неравновесному характеру хим. процессов в верх. слоях атмосферы при очень небольшом числе элементов возникает необычайное многообразие наблюдаемых прир. явлений.
Неравновесные электронно-возбужденные состояния молекул играют решающую роль в первичных актах фотосинтеза. Кванты света поглощаются системой молекул хлорофилла, затем по экситонному механизму энергия возбуждения передается димеру хлорофилла с послед. фотохим. разделением заряда. Порождаемые внеш. воздействием (светом, хим. превращениями в среде) неравновесно возбужденные атомы, молекулы, сложные мол. комплексы обусловливают высокую избирательность биохим. р-ций, управление и самоорганизацию хим., биол. и физиол. процессов, характерных для живой природы (см. Самоорганизация в неравновесных процессах).
Лит.: Кондратьев В. Н., Никитин Е.Е., Кинетика и механизм газофазных реакций, М., 1974; Трое Ю., Вагнер X., в кн.: Физическая химия быстрых реакций, пер. с англ., М., 1976, с. 13-105; Полак Л. С., Неравновесная химическая кинетика и ее применение, М., 1979; Гордиец Б.Ф., Оси-пов А. И., Шелепин Л. А., Кинетические процессы в газах и молекулярные лазеры, М., 1980; Кузнецов Н. М., Кинетика мономолекулярных реакций, М., 1982; Крылов О. В., "Кинетика и катализ", 1989, т. 30, вып. 3, с. 519-32.
Н. М. Кузнецов.