Энтропия
ЭНТРОПИЯ, ф-ция состояния S термодинамич. системы, изменение к-рой dS для бесконечно малого обратимого изменения состояния системы равно отношению кол-ва теплоты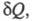 полученного системой в этом процессе (или отнятого от системы), к абс. т-ре Т:
полученного системой в этом процессе (или отнятого от системы), к абс. т-ре Т:
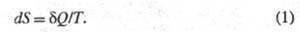
Величина dS является полным дифференциалом, т. е. ее интегрирование по любому произвольно выбранному пути дает разность между значениями энтропии в начальном (А) и конечном (В) состояниях:
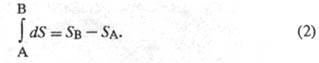
Теплота не является ф-цией состояния, поэтому интеграл от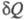 зависит от выбранного пути перехода между состояниями А и В.
зависит от выбранного пути перехода между состояниями А и В.
Энтропия измеряется в Дж/(моль-град).
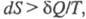 для вторых
для вторых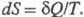
Энтропия как ф-ция внутренней энергии U системы, объема V и числа молей ni i-го компонента представляет собой характеристич. ф-цию (см. Термодинамические потенциалы). Это является следствием первого и второго начал термодинамики и записывается ур-нием:
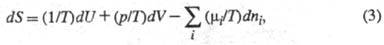
где р - давление;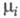 - хим. потенциал i-го компонента. Производные энтропии по естеств. переменным U, V и ni равны:
- хим. потенциал i-го компонента. Производные энтропии по естеств. переменным U, V и ni равны:
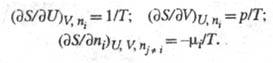
Простые ф-лы связывают энтропию с теплоемкостями при пост. давлении Ср и пост, объеме Cv:
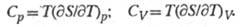
С помощью энтропии формулируются условия достижения термодинамич. равновесия системы при постоянстве ее внутр. энергии, объема и числа молей i-го компонента (изолир. система) и условие устойчивости такого равновесия:
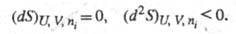
Это означает, что энтропия изолир. системы достигает максимума в состоянии термодинамич. равновесия. Самопроизвольные процессы в системе могут протекать только в направлении возрастания энтропии.
Энтропия относится к группе термодинамич. ф-ций, называемых ф-циями Массье-Планка. Другие ф-ции, принадлежащие к этой группе - ф-ция Массье Ф1 = S - (1/T)U и ф-ция Планка Ф2 = S - (1/T)U — (p/T)V, м. б. получены в результате применения к энтропии преобразования Лежандра.
Согласно третьему началу термодинамики (см. Тепловая теорема), изменение энтропии в обратимой хим. р-ции между в-вами в конденсир. состоянии, стремится к нулю при T 0:
0:
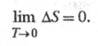
Постулат Планка (альтернативная формулировка тепловой теоремы) устанавливает, что энтропия любого хим. соед. в конденсир. состоянии при абс. нуле т-ры является условно нулевой и м. б. принята за начало отсчета при определении абс. значения энтропии в-ва при любой т-ре. Ур-ния (1) и (2) определяют энтропию с точностью до постоянного слагаемого.
В хим. термодинамике широко используют след. понятия: стандартная энтропия 5ю, т. е. энтропия при давлении р=1,01 x 105 Па (1 атм); стандартная энтропия хим. р-ции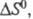 т. е. разница стандартных энтропий продуктов и реагентов; парциальная молярная энтропия компонента многокомпонентной системы
т. е. разница стандартных энтропий продуктов и реагентов; парциальная молярная энтропия компонента многокомпонентной системы
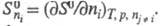
Для расчета хим. равновесий применяют ф-лу:
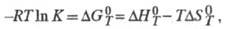
где К - константа равновесия;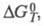
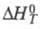 и
и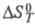 - соотв. стандартные энергия Гиббса, энтальпия и энтропия р-ции; R -газовая постоянная.
- соотв. стандартные энергия Гиббса, энтальпия и энтропия р-ции; R -газовая постоянная.
Определение понятия энтропии для неравновесной системы опирается на представление о локальном термодинамич. равновесии. Локальное равновесие подразумевает выполнение ур-ния (3) для малых объемов неравновесной в целом системы (см. Термодинамика необратимых процессов). При необратимых процессах в системе может осуществляться производство (возникновение) энтропии. Полный дифференциал энтропии определяется в этом случае неравенством Карно-Клаузиуса:
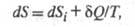
где dSi > 0 - дифференциал энтропии, не связанный с потоком тепла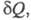 а обусловленный производством энтропии за счет необратимых процессов в системе (диффузии, теплопроводности, хим. р-ций и т. п.). Локальное производство энтропии
а обусловленный производством энтропии за счет необратимых процессов в системе (диффузии, теплопроводности, хим. р-ций и т. п.). Локальное производство энтропии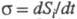 (t - время) представляется в виде суммы произведений обобщенных термодинамич. сил Xi на обобщенные термодинамич. потоки Ji:
(t - время) представляется в виде суммы произведений обобщенных термодинамич. сил Xi на обобщенные термодинамич. потоки Ji:
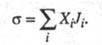
Производство энтропии за счет, напр., диффузии компонента i обусловлено силой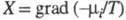 и потоком в-ва J; производство энтропии за счет хим. р-ции- силой Х=А/Т, где А-химическое сродство, и потоком J, равным скорости р-ции. В статистич. термодинамике энтропия изолир. системы определяется соотношением:
и потоком в-ва J; производство энтропии за счет хим. р-ции- силой Х=А/Т, где А-химическое сродство, и потоком J, равным скорости р-ции. В статистич. термодинамике энтропия изолир. системы определяется соотношением: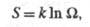 где k - постоянная Больцмана;
где k - постоянная Больцмана;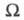 - термодинамич. вес состояния, равный числу возможных квантовых состояний системы с заданными значениями энергии, объема, числа частиц. Равновесное состояние системы отвечает равенству заселенностей единичных (невырожденных) квантовых состояний. Возрастание энтропии при необратимых процессах связано с установлением более вероятного распределения заданной энергии системы по отд. подсистемам. Обобщенное статистич. определение энтропии, относящееся и к неизолир. системам, связывает энтропию с вероятностями разл. микросостояний след. образом:
- термодинамич. вес состояния, равный числу возможных квантовых состояний системы с заданными значениями энергии, объема, числа частиц. Равновесное состояние системы отвечает равенству заселенностей единичных (невырожденных) квантовых состояний. Возрастание энтропии при необратимых процессах связано с установлением более вероятного распределения заданной энергии системы по отд. подсистемам. Обобщенное статистич. определение энтропии, относящееся и к неизолир. системам, связывает энтропию с вероятностями разл. микросостояний след. образом:
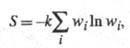
где wi - вероятность i-го состояния.
Абсолютную энтропию хим. соед. определяют экспериментально, гл. обр. калориметрич. методом, исходя из соотношения:
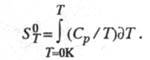
Использование второго начала позволяет определять энтропию хим. р-ций по эксперим. данным (метод электродвижущих сил, метод давления пара и др.). Возможен расчет энтропии хим. соед. методами статистич. термодинамики, исходя из мол. постоянных, мол. массы, геометрии молекулы, частоты нормальных колебаний. Такой подход успешно осуществляется для идеальных газов. Для конденсир. фаз статистич. расчет дает значительно меньшую точность и проводится в ограниченном числе случаев; в последние годы в этой области достигнуты значит. успехи.
Энтропии хим. соед. табулированы в справочниках. Как правило, для конденсир. фаз приводят результаты калориметрич. измерений, для газообразных - статистич. расчета.
Первая мат. формулировка второго начала термодинамики принадлежит Р. Клаузиусу (1854), к-рый ввел понятие энтропии в 1865; связь энтропии с вероятностью состояния системы впервые была установлена Л. Больцманом в 1872.
Лит. см. при ст. Химическая термодинамика.
М. В. Коробов.