2.4. Приближение супермолекулы
С точки зрения построения модели наиболее простым способом учета сольватации является включение большого числа молекул среды в систему, для которой проводится квантовохимический расчет. Все ее электроны (сольватируемой молекулы и молекул среды) включаются в электронный гамильтониан. Этот способ является непосредственным обобщением квантовохимических методов, развитых для отдельных молекул на случай больших систем, состоящих из нескольких или даже большого числа отдельных молекул. Если нам удастся учесть таким способом взаимодействие растворенной молекулы с большим числом молекул cреды, рассчитать энергетически наиболее выгодную конформацию растворенной молекулы и конфигурацию молекул растворителя и получить для этой конфигурации электронную волновую функцию, то мы сможем объяснить или даже предсказать практически все интересующие нас свойства молекулы в растворе. При этом следует с особой осторожностью подходить к выбору квантовохимического метода, к которому в этом случае предъявляются повышенные требования, а именно он должен быть пригоден для изучения межмолекулярных взаимодействий.
Из предыдущей главы было видно, что многие квантовохимические методы, которые успешно используются для изучения реакционной способности органических соединений дают неправильные результаты при расчете параметров, характеризующих межмолекулярные взаимодействия. Например, метод МПДП дает неудовлетворительные результаты при расчете систем с водородными связями. Поэтому при использовании приближения "супермолекулы" для учета сольватации приходится выбирать достаточно совершенный квантовохимический метод. При этом возникают трудности, связанные с очень большим размером супермолекулы, которая состоит из растворенного соединения и молекул среды. Кроме того, в супермолекуле обычно неизвестно большое число геометрических параметров, которые определяют строение сольватационной оболочки. Поэтому выполнить реально такие расчеты удается только для систем с небольшим числом молекул среды.
Такой учет сольватации, при котором система из сольватируемой молекулы и некоторого ограниченного числа молекул растворителя рассчитывается квантовохимическим методом как одна молекула, получил название "приближения супермолекулы" [93, 94]. Расчеты в этом приближении широко распространены. К ним относятся работы, в которых вычисляются параметры для комплексов, состоящих из растворенной молекулы и одной молекулы растворителя, а также расчеты для комплексов с двумя, тремя и т.д. молекулами среды. В этих работах впервые удалось получить данные о строении и энергии взаимодействия для гидратных оболочек ионов ОН- и Н3О+ [65]. Несколько позже аналогичными методами исследованы гидратационные оболочки простейших ионов: Li+, Be2+, Na+, Mg2+, Al3+, К+, Ca2+, F-, Сl-, NH4+ [95 - 102], CH5+, CH5- [103], алкиламмониевых ионов [104].
Методика проведения перечисленных выше работ заложила основу подхода к изучению сольватации в приближении супермолекулы. Суть этих подходов заключается в следующем. Вначале рассчитывается энергия взаимодействия иона с одной молекулой воды и определяется наиболее энергетически выгодная конформация, потом в систему добавляется еще одна молекула воды и вновь рассчитываются энергия взаимодействия и структура комплекса и т.д. В результате такого расчета получается набор величин, которые являются энергиями гидратации иона А каждой последующей молекулой воды (En,n-1):
А•(n - 1)Н2O+ Н2O → А•nН2O+ En,n-1.
Для простейших ионов эти энергии были сопоставлены с экспериментальными данными, полученными методами масс-спектрометрии высоких давлений и циклотронного резонанса. Хорошее согласие с экспериментом подтвердило широкие возможности квантовохимических расчетов для изучения сольватации. Кроме того, были получены данные о числе молекул воды в первой гидратационной сфере и их конфигурация.
При переходе к более сложным молекулам расчеты в приближении супермолекулы существенно усложняются, так как центров сольватации уже много и заполнить даже всю первую гидратационную оболочку практически не удается. Поэтому в большинстве работ придерживаются следующей последовательности:
1) детально изучается взаимодействие сольватируемой молекулы с одной молекулой растворителя и получается предварительное представление о строении сольватационной оболочки;
2) изучается влияние второй, третьей и т.д. молекул растворителя на строение сольватационной оболочки и уточняются данные, полученные на первом этапе;
3) в изучаемую систему вводится максимально возможное количество молекул растворителя, для которого удается выполнить расчет, и для такой супермолекулы вычисляются все интересующие величины.
Детальное изучение взаимодействия сольватируемого соединения и одной молекулы растворителя включает их сближение (с различной взаимной ориентацией) и вращение вокруг собственных локальных осей. На этом этапе получают данные об основных наиболее выгодных положениях молекул растворителя, энергиях взаимодействия в таких положениях, расстояниях между растворенным соединением и молекулами растворителя и о подвижности последних. Обычно различают три вида подвижности: в положении равновесия, вблизи положения равновесия и вдали от положения равновесия.
Подвижность в положении равновесия — возможность молекулы растворителя вращаться вокруг собственных локальных осей. Для изучения этого вида подвижности рассчитывают зависимость энергии взаимодействия растворенного соединения и молекулы растворителя от углов поворота последней вокруг ее локальных осей. Расстояние между ними при этом не меняется. Подвижность вблизи положения равновесия — возможность молекулы растворителя смещаться на небольшие расстояния от положения равновесия. Для изучения этого вида подвижности рассчитывают форму ППЭ при небольших удалениях молекулы растворителя от положения равновесия без разрыва водородных связей, которые были в положении равновесия. Подвижность вдали от положения равновесия — возможность молекулы растворителя смещаться на большие расстояния от положения равновесия. Для изучения этого вида подвижности рассчитывают ППЭ при больших удалениях молекулы растворителя от положения равновесия, когда водородные связи между ней и растворенным соединением разорваны.
На втором этапе увеличивают число молекул растворителя в супермолекуле. При этом используют данные о положениях локальных минимумов, полученные на первом этапе, и лишь уточняют их положение. На третьем этапе в супермолекулу включают максимально возможное количество молекул растворителя (исходя из возможностей ЭВМ) и проводят расчет того или иного параметра.
В качестве примера рассмотрим результаты работы [105], в которой изучено строение гидратационной оболочки мочевины. Это соединение обладает рядом уникальных свойств. Так, в его присутствии резко снижается температура, при которой в водных растворах происходит денатурация белков и других биополимеров, повышается растворимость неэлектролитов и т.д. Принято говорить, что в присутствии мочевины разрушаются водородные связи между молекулами воды, другими словами, молекулы воды в водном растворе становятся более подвижными. Это приводит к перечисленным выше химическим и биохимическим эффектам. Однако механизм этого явления на микроскопическом уровне установлен не был. Для решения этого вопроса было изучено [105] строение гидратационной оболочки мочевины с помощью квантовохимических расчетов методами ППДП/2 и ОСТ-ЗГФ.
На первом этапе был проведен расчет строения комплекса, образованного одной молекулой мочевины с одной молекулой воды, и рассмотрена подвижность последней в этом комплексе. Оказалось, что наиболее устойчивый комплекс имеет следующую конфигурацию:
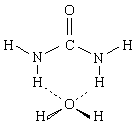
В этом комплексе молекула воды связана двумя водородными связями с двумя атомами Н связей N—Н, которые находятся в транс-положении по отношению к карбонильной группе. Расчет также показал, что в этом положении молекула воды обладает необычайно высокой подвижностью. Она может смещаться на расстояния до 0,15—0,20 нм от положения равновесия, и при этом энергия взаимодействия в комплексе почти не меняется. Таким образом, было обнаружено, что в исследованном комплексе молекулы воды и мочевины частично сохраняют свободу независимых перемещений в пространстве.
На втором этапе было увеличено количество молекул воды в комплексе, однако оказалось, что вторая молекула Н2О не может связаться с молекулой мочевины аналогично первой и образовать комплекс типа
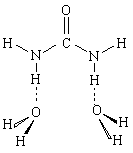
Таким образом, первая молекула воды, присоединяясь к мочевине, попадает в широкую потенциальную яму (именно этим объясняется ее высокая подвижность), но вторая молекула воды в эту потенциальную яму попасть не может, поэтому при увеличении количества молекул воды в комплексе высокая подвижность одной из них сохраняется. По-видимому, наличие этой достаточно широкой и глубокой потенциальной ямы обусловливает способность мочевины "разрушать структуру воды".
Введение мочевины в воду приводит к тому, что часть молекул воды, которые в чистом водном растворе (без мочевины) образовывали упорядоченную структуру (за счет образования водородных связей между собой), попадают в широкую потенциальную яму, созданную мочевиной. Такие молекулы не занимают определенного положения в пространстве и сохраняют способность перемещаться на большие расстояния (в пределах размеров потенциальной ямы). Поэтому как сами эти молекулы, так и молекулы воды из их непосредственного окружения не могут участвовать в образовании упорядоченной структуры воды. Подвижность молекул воды в данном случае увеличивается. Происходит как бы локальное повышение термодинамической температуры. Однако в действительности этот эффект обусловлен не повышением энергии молекулы воды, локализованной на ее вращательных и поступательных степенях свободы, а снижением потенциальных барьеров на пути ее перемещения.
Таким образом, предложен [105] механизм повышения термодинамической температуры водных растворов при добавлении в них мочевины. Квантовохимические расчеты помогли его найти, но сами по себе они не могут служить доказательством, что экспериментально наблюдаемые эффекты происходят именно по такому механизму. Для этого необходимо на основе предложенного механизма сделать предсказания, которые можно проверить экспериментально. Такие предсказания были сделаны [105]. Они касались влияния алкильных заместителей на способность мочевины повышать термодинамическую температуру водных растворов.
У метилмочевины наиболее устойчивой является следующая конформация:
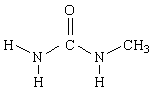
Наличие двух связей N—Н в транс-положении по отношению к карбонильной группе ведет к сохранению особенностей строения комплекса, описанных выше. Поэтому у метилмочевины сохраняется способность повышать термодинамическую температуру воды.
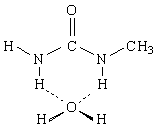
У диметилмочевины возможны два изомера
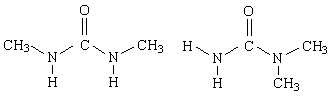
У первого из них две связи N—Н в транс-положении по отношению к карбонильной группе, а у второго - лишь одна. Следовательно, первый изомер будет повышать термодинамическую температуру воды и приводить к тем же эффектам, что и мочевина, а второй - нет. Это теоретическое предсказание получило экспериментальное подтверждение.
Приближение супермолекулы позволяет решать многие задачи, связанные с влиянием сольватации на реакционную способность органических соединений. Однако при его использовании необходимо самое серьезное внимание уделить выбору метода расчета. При этом следует руководствоваться двумя требованиями: 1) метод расчета должен быть достаточно точным и хорошо передавать как основные свойства растворенной молекулы, так и строение сольватационных оболочек; 2) расчет в приближении супермолекулы связан с вычислением электронной волновой функции для очень большой системы, и он должен быть практически реализуем исходя из возможностей имеющихся ЭВМ, их быстродействия и памяти. До недавнего времени расчеты с учетом сольватации в приближении супермолекулы проводились методами ППДП/2 и ОСТ-ЗГФ. Именно эти методы были использованы в приведенном выше примере изучения особенностей строения гидратационной оболочки мочевины. В настоящее время предпочтительнее использовать метод МПДП/Н и неэмпирические расчеты в базисе 3-21ГФ.