Конфигурациoнного взаимодействия метод
КОНФИГУРАЦИOННОГО ВЗАИМОДЕЙСТВИЯ МЕТОД (метод взаимодействия конфигураций), квантовохим. метод приближенного решения ур-ния Шрёдингера для многоэлектронной мол. системы в основном и возбужденных состояниях. Основан на адиабатическом приближении и позволяет в принципе находить электронные волновые ф-ции и энергетич. уровни молекулы с любой наперед заданной точностью, чем отличается от заведомо приближенных молекулярных орбиталей методов. Осн. понятие конфигурационного взаимодействия метода - конфигурац. ф-ция состояния (КФС) -приближенная волновая ф-ция молекулы для заданного электронного состояния, определяемая на основе метода мол. орбиталей как антисимметризованное произведение волновых ф-ций отдельных электронов, составленное с учетом суммарного спина, принципа Паули и симметрии расположения ядер. КФС отвечает определенному распределению электронов по орбиталям, т. е. определенной электронной конфигурации, и передает особенности волновой ф-ции молекулы лишь в той мере, в какой кулоновское взаимод. всех электронов можно приближенно рассматривать как взаимод. электрона с усредненным полем. Взаимная согласованность движений электронов (электронная корреляция) не описывается одной КФС, однако состояние молекулы можно охарактеризовать неск. КФС, каждая из к-рых выделяет одну из особенностей сложного движения электронов. Напр., в электронном распределении, описывающем хим. связь, одни КФС могут выделять ковалентные, а другие - ионные составляющие связи (см. Валентных связей метод). Сущность конфигурационного взаимодействия метода представление сложного согласованного движения электронов в многоэлектронной мол. системе комбинацией (суперпозицией) относительно независимых движений, к-рым отвечают КФС. Конфигурационного взаимодействия метод является линейным вариационным методом, в к-ром каждое электронное состояние приближенно описывается волновой ф-цией y, представленной линейной комбинацией конечного числа т специальным образом выбранных КФС Фk: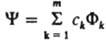 , где ck - подлежащие определению коэффициенты, отражающие роль отд. типов движении электронов в эволюции системы как целого.
, где ck - подлежащие определению коэффициенты, отражающие роль отд. типов движении электронов в эволюции системы как целого. 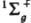 , т. е. состояния с нулевым суммарным спином и волновыми ф-циями, не меняющимися при всех операциях симметрии системы ядер. В рамках метода мол. орбиталей эти состояния можно описать двумя КФС Ф1 и Ф2, соответствующими электронным конфигурациям sg2 и su2 (мол. орбитали sg и su симметричны относительно оси, соединяющей ядра, и сохраняют или меняют знак при инверсии). В рамках конфигурационного взаимодействия метода указанные состояния описываются более точно волновыми ф-циями y1=с1Ф1+с2Ф2 и y2=-с2Ф1+C1Ф2, где c1 и с2 - подлежащие определению коэффициенты, удовлетворяющие нормировочному условию: с12+с22=1. Среднее значение энергии молекулы для состояния с ф-цией y1 ниже, чем для Ф1 т.к. при учете межэлектронного отталкивания уменьшается вероятность локализации обоих электронов в одном малом объеме и увеличивается среднее расстояние между электронами. Для одной и той же молекулы в зависимости от расположения ядер и типа состояния вклады разл. КФС в волновую ф-цию могут меняться, поэтому в одних случаях можно ограничиться единственной КФС, применяя метод мол. орбиталей, а в других необходимо учитывать многоконфигурац. характер волновой ф-ции, т.е. использовать конфигурационного взаимодействия метод. Качеств, заключения о необходимости применения конфигурационного взаимодействия метода часто дает анализ корреляц. диаграмм (см. Орбиталь, Вудворда-Хофмана правила). Напр., основное состояние молекулы Н2 вблизи равновесного межъядерного расстояния хорошо описывается методом мол. орбиталей, т. к. Y1~Ф1. Однако вблизи диссоциац. предела необходим учет электронной корреляции с помощью конфигурационного взаимодействия метода, т.к.
, т. е. состояния с нулевым суммарным спином и волновыми ф-циями, не меняющимися при всех операциях симметрии системы ядер. В рамках метода мол. орбиталей эти состояния можно описать двумя КФС Ф1 и Ф2, соответствующими электронным конфигурациям sg2 и su2 (мол. орбитали sg и su симметричны относительно оси, соединяющей ядра, и сохраняют или меняют знак при инверсии). В рамках конфигурационного взаимодействия метода указанные состояния описываются более точно волновыми ф-циями y1=с1Ф1+с2Ф2 и y2=-с2Ф1+C1Ф2, где c1 и с2 - подлежащие определению коэффициенты, удовлетворяющие нормировочному условию: с12+с22=1. Среднее значение энергии молекулы для состояния с ф-цией y1 ниже, чем для Ф1 т.к. при учете межэлектронного отталкивания уменьшается вероятность локализации обоих электронов в одном малом объеме и увеличивается среднее расстояние между электронами. Для одной и той же молекулы в зависимости от расположения ядер и типа состояния вклады разл. КФС в волновую ф-цию могут меняться, поэтому в одних случаях можно ограничиться единственной КФС, применяя метод мол. орбиталей, а в других необходимо учитывать многоконфигурац. характер волновой ф-ции, т.е. использовать конфигурационного взаимодействия метод. Качеств, заключения о необходимости применения конфигурационного взаимодействия метода часто дает анализ корреляц. диаграмм (см. Орбиталь, Вудворда-Хофмана правила). Напр., основное состояние молекулы Н2 вблизи равновесного межъядерного расстояния хорошо описывается методом мол. орбиталей, т. к. Y1~Ф1. Однако вблизи диссоциац. предела необходим учет электронной корреляции с помощью конфигурационного взаимодействия метода, т.к. 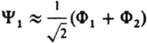 .
. Как правило, вблизи равновесного расположения ядер достаточно в основном состоянии применять метод мол. орбиталей. Если же при изменении положений ядер происходит разрыв или образование связей, то без учета электронной корреляции нельзя получить правильное описание процесса. В квантовохим. задачах применяются разл. варианты конфигурационного взаимодействия метода, отличающиеся способом выбора учитываемых КФС. Нередко совмещают конфигурационного взаимодействия метод с методами возмущений теории, что позволяет учесть вклады от целых классов КФС в полную волновую ф-цию. Разработаны компромиссные варианты конфигурационного взаимодействия метода, в к-рых описывается лишь наиб. важная часть корреляции, отвечающая взаимной обусловленности движений электронных пар (методы связанных электронных пар, кластерных разложений и др.). Использование конфигурационного взаимодействия метода определяется той ролью, к-рую играет электронная корреляция в мол. процессах. Учет корреляции необходим при описании дисперсионного взаимодействия, изменения фотоэлектронных и Оже-спектров при изменении структурных фрагментов молекулы. Во мн. хим. р-циях, в т.ч. каталитических, волновые ф-ции переходных состоянии имеют существенно многоконфигурац. характер; то же относится к возбужденным состояниям молекул. С электронной корреляцией связывают нарушения Хунда правил, изменение порядка заполнения одноэлектронных уровней в атомах переходных элементов. По мере развития представления о природе хим. связи необходимость учета электронной корреляции методами типа конфигурационного взаимодействия метода приобретает все более важное значение. Методы, близкие к конфигурационного взаимодействия методу, все шире используются при решении задач о колебаниях многоатомных молекул.
Литература
Рамбидн Н.Г., Степанов Н.Ф., Дементьев А. И., Квантово-механнческие расчеты двухатомных молекул, М., 1979 (Итоги науки и техники. Сер. Строение молекул и химическая связь, т. 7); Уилсон С., Электронные корреляции в молекулах, пер. с англ., М., 1987. В. И. Пупышев.
Ещё по теме
1.3. Расчет индексов реакционной способности
Электронная корреляция в химии
Квантово-химические методы в химии — применение и точность расчётов
Квантовохимические расчёты электронных переходов
Квантово-механические методы расчёта молекул
Спин-орбитальное взаимодействие в квантовой физике
Метод Хюккеля в квантовой химии
Обменное взаимодействие в квантовой механике
p-Электронное приближение в квантовой химии
Методы расчёта строения молекул в электронно-возбуждённых состояниях
Ядерный квадрупольный резонанс — принципы и применение