Электронное приближение
-ЭЛЕКТРОННОЕ ПРИБЛИЖЕНИЕ, квантовохим. метод изучения энергетич. состояний ненасыщенных соед., в к-ром св-ва молекулы соотносятся со строением системы -орбита-лей. В рамках молекулярных орбиталей методов все орбитали молекулы, ядерная конфигурация к-рой имеет плоскость симметрии, можно разделить на
-орбита-лей. В рамках молекулярных орбиталей методов все орбитали молекулы, ядерная конфигурация к-рой имеет плоскость симметрии, можно разделить на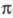 -орбитали, меняющие знак при отражении в плоскости симметрии, и
-орбитали, меняющие знак при отражении в плоскости симметрии, и -орбитали, не меняющие знака. Для многих классов соед., напр. ненасыщенных углеводородов, высшие занятые и низшие виртуальные (незанятые) орбитали относятся к
-орбитали, не меняющие знака. Для многих классов соед., напр. ненасыщенных углеводородов, высшие занятые и низшие виртуальные (незанятые) орбитали относятся к -типу, а усредненное поле, создаваемое
-типу, а усредненное поле, создаваемое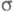 -электронами, можно считать локально постоянным. Это позволяет пренебречь взаимным влиянием
-электронами, можно считать локально постоянным. Это позволяет пренебречь взаимным влиянием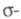 и
и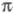 -электронных подсистем при изучении низколежащих по энергии электронных состояний.
-электронных подсистем при изучении низколежащих по энергии электронных состояний.
Для молекул, обладающих плоскостью симметрии, выделение - и
- и -орбиталей не составляет трудностей. Напр., для плоской молекулы этилена
-орбиталей не составляет трудностей. Напр., для плоской молекулы этилена -орбитали считаются построенными из гибридных sp2-орбиталей атома С и 1s-орбиталей атомов Н, а
-орбитали считаются построенными из гибридных sp2-орбиталей атома С и 1s-орбиталей атомов Н, а -орбитали - из негибридизированных 2р-орби-талей атомов С. Для молекул, не имеющих плоскости симметрии, строгое выделение системы
-орбитали - из негибридизированных 2р-орби-талей атомов С. Для молекул, не имеющих плоскости симметрии, строгое выделение системы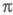 -орбиталей невозможно. В этом случае для
-орбиталей невозможно. В этом случае для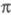 -электронного приближения используют такие орбитали, к-рые более всего напоминают р-орбитали атомов. Напр., для пропилена
-электронного приближения используют такие орбитали, к-рые более всего напоминают р-орбитали атомов. Напр., для пропилена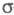 -орбитали можно описать с помощью тех же орбиталей, что и в случае этилена, и sp3-орбиталей атома С метильной группы. В качестве p-ороиталей рассматривают негибридизированные 2p-орбитали атомов С.
-орбитали можно описать с помощью тех же орбиталей, что и в случае этилена, и sp3-орбиталей атома С метильной группы. В качестве p-ороиталей рассматривают негибридизированные 2p-орбитали атомов С.
Мол. системы, для к-рых возможно разделение орбиталей на и
и -орбитали, наз. часто
-орбитали, наз. часто -электронными системами или просто
-электронными системами или просто -системами. Как правило,
-системами. Как правило,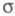 -орбитали носят локализованный характер, обычно они двухцентровые.
-орбитали носят локализованный характер, обычно они двухцентровые. -Орбитали существенно менее локализованы и могут иметь трех-, четырех- или многоцентровый характер, что в химии связывают с понятием сопряжения связей (бутадиен, бензол и т. п.). При изменении строения молекулы (напр., введении заместителей) изменения св-в связывают именно с
-Орбитали существенно менее локализованы и могут иметь трех-, четырех- или многоцентровый характер, что в химии связывают с понятием сопряжения связей (бутадиен, бензол и т. п.). При изменении строения молекулы (напр., введении заместителей) изменения св-в связывают именно с -орбиталями, а изменением
-орбиталями, а изменением -орбиталей пренебрегают. Взаимное влияние
-орбиталей пренебрегают. Взаимное влияние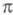 -орбиталей и остальных мол. орбиталей учитывают как изменение св-в молекулы при введении заместителя (поляризацию) или как дополнит. сопряжение, используя методы, напр., возмущений теории. Надежность результатов, получаемых в p-электронном приближении, определяется тем, насколько удалены друг от друга локализованные мол. орбитали, взаимодействующие с системой
-орбиталей и остальных мол. орбиталей учитывают как изменение св-в молекулы при введении заместителя (поляризацию) или как дополнит. сопряжение, используя методы, напр., возмущений теории. Надежность результатов, получаемых в p-электронном приближении, определяется тем, насколько удалены друг от друга локализованные мол. орбитали, взаимодействующие с системой -орбиталей, а также тем, насколько различаются эти мол. орбитали по энергии.
-орбиталей, а также тем, насколько различаются эти мол. орбитали по энергии.
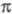 -электронном приближении для расчетов основного состояния молекулы используют полуэмпирические методы квантовой химии, основанные на пренебрежении дифференциальным перекрыванием атомных орбиталей. Расчет низколежащих возбужденных состояний возможен с использованием конфигурационного взаимодействия метода в том его варианте, в к-ром однократно возбужденные электронные конфигурации получают заменой занятых
-электронном приближении для расчетов основного состояния молекулы используют полуэмпирические методы квантовой химии, основанные на пренебрежении дифференциальным перекрыванием атомных орбиталей. Расчет низколежащих возбужденных состояний возможен с использованием конфигурационного взаимодействия метода в том его варианте, в к-ром однократно возбужденные электронные конфигурации получают заменой занятых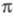 -орбиталей на аналогичные вакантные. К таким методам относится, напр., метод Паризе ра-Парра-Попла (метод ППП). Взаимодействие
-орбиталей на аналогичные вакантные. К таким методам относится, напр., метод Паризе ра-Парра-Попла (метод ППП). Взаимодействие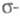 и
и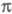 -орбиталей проявляется как зависимость молекулярных интегралов и геометрии молекулы от
-орбиталей проявляется как зависимость молекулярных интегралов и геометрии молекулы от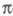 -электронной плотности.
-электронной плотности. В связи с развитием расчетных методов
 -электронное приближение вытесняется полуэмпирич. методами без выделения
-электронное приближение вытесняется полуэмпирич. методами без выделения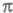 -системы или неэмпирическими методами. Однако для мол. систем большого размера (полиацетилены, поликонденсиров. системы) учет всех взаимод. затруднен. Для них обычно используют упрощенные варианты
-системы или неэмпирическими методами. Однако для мол. систем большого размера (полиацетилены, поликонденсиров. системы) учет всех взаимод. затруднен. Для них обычно используют упрощенные варианты -электронного приближения. Напр., в т. наз. приближении Хаббарда ненулевыми мол. интегралами считаются лишь одноцентровые интегралы межэлектронного отталкивания. Еще более простым является приближение Хюккеля, в к-ром двухэлектронными взаимод. вообще пренебрегают, а в матрице эффективного гамильтониана, определяющего
-электронного приближения. Напр., в т. наз. приближении Хаббарда ненулевыми мол. интегралами считаются лишь одноцентровые интегралы межэлектронного отталкивания. Еще более простым является приближение Хюккеля, в к-ром двухэлектронными взаимод. вообще пренебрегают, а в матрице эффективного гамильтониана, определяющего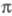 -орбиталь, сохраняются лишь диагональные элементы и те из недиагональных, к-рые можно соотнести с валентным штрихом в структурной ф-ле молекулы (см. Хюккеля метод).
-орбиталь, сохраняются лишь диагональные элементы и те из недиагональных, к-рые можно соотнести с валентным штрихом в структурной ф-ле молекулы (см. Хюккеля метод). Упомянутые упрощенные варианты
 -электронного приближения предполагают определение полуэмпирич. параметров по результатам расчета энергии
-электронного приближения предполагают определение полуэмпирич. параметров по результатам расчета энергии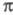 -системы, потенциалов ионизации или энергий возбуждения простых молекул (без заместителей).
-системы, потенциалов ионизации или энергий возбуждения простых молекул (без заместителей). -Электронное приближение служит для описания наиб. изменчивой, легко поляризуемой и относительно независимой части электронного распределения молекулы. Корреляционные соотношения позволяют соотнести энергию
-Электронное приближение служит для описания наиб. изменчивой, легко поляризуемой и относительно независимой части электронного распределения молекулы. Корреляционные соотношения позволяют соотнести энергию -системы и распределение электронной плотности с хим. св-вами молекулы.
-системы и распределение электронной плотности с хим. св-вами молекулы. У многих мол. систем среди низших возбужденных состояний есть такие, для описания к-рых необходим учет
 -возбуждений, напр. если в
-возбуждений, напр. если в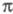 -систему включается гетероатом с неподеленными парами электронов. В таких случаях
-систему включается гетероатом с неподеленными парами электронов. В таких случаях -электронное приближение
-электронное приближение становится слишком грубым. То же можно утверждать и для мол. систем с заметным переносом заряда при возбуждении. У молекул, традиционно относимых к
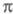 -системам, высшие занятые
-системам, высшие занятые -орбитали могут лежать по энергии выше низших занятых
-орбитали могут лежать по энергии выше низших занятых -орбиталей (пример - бензол). При внеш. воздействии на такие молекулы необходим учет изменения не только
-орбиталей (пример - бензол). При внеш. воздействии на такие молекулы необходим учет изменения не только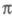 -орбиталей, но и
-орбиталей, но и -орбиталей (напр., при анализе магн. восприимчивости).
-орбиталей (напр., при анализе магн. восприимчивости). С 80-х гг. стали заметны две тенденции в развитии методов изучения
 -электронных систем. Первая - отказ от учета особой роли
-электронных систем. Первая - отказ от учета особой роли -орбиталей и анализ молекулы как целого или же выделение группы низколежащих орбиталей (или групп орбиталей) молекулы независимо от типа их симметрии. Вторая тенденция - переход к чисто топологич. моделированию высших занятых и низших виртуальных мол. орбиталей; топологич. моделирование связано с методами типа метода Хюккеля и теориями реакционной способности, основанными на корреляционных соотношениях или принципах сохранения орбитальной симметрии (см. Вудворда-Хофмана правила).
-орбиталей и анализ молекулы как целого или же выделение группы низколежащих орбиталей (или групп орбиталей) молекулы независимо от типа их симметрии. Вторая тенденция - переход к чисто топологич. моделированию высших занятых и низших виртуальных мол. орбиталей; топологич. моделирование связано с методами типа метода Хюккеля и теориями реакционной способности, основанными на корреляционных соотношениях или принципах сохранения орбитальной симметрии (см. Вудворда-Хофмана правила). Лит.: Дьюар М., Теория молекулярных орбиталей в органической химии, пер. с англ., М., 1972; Полуэмпирические методы расчета электронной структуры, пер. с англ., т. 1-2, М., 1980; Травень В. Ф., Электронная структура и свойства органических молекул, М., 1989.
В. И. Пупышев.