Органическая химия
ОРГАНИЧЕСКАЯ ХИМИЯ, наука, изучающая соединения углерода с др. элементами (органические соединения), а также законы их превращений. Назв. "органическая химия" возникло на ранней стадии развития науки, когда предмет изучения ограничивался соед. углерода растит, и животного происхождения. Не все соед. углерода классифицируются как органические. Напр., СО2, HCN, CS2 традиционно относят к неорганическим. Условно можно считать, что прототипом орг. соед. является метан СН4.
К настоящему времени число известных орг. соед. превышает 10 млн. и увеличивается каждый год на 250-300 тыс. Многообразие орг. соед. определяется уникальной способностью атомов углерода соединяться друг с другом простыми и кратными связями, образовывать соед. с практически неогранич. числом атомов, связанных в цепи, циклы, бициклы, трициклы, полициклы, каркасы и др., образовывать прочные связи почти со всеми элементами периодич. системы, а также явлением изомерии-существованием разных по св-вам в-в, обладающих одним и тем же составом и мол. массой.
Многообразие и громадное число орг. соед. определяет значение органической химии как крупнейшего раздела совр. химии. Окружающий нас мир построен гл. обр. из орг. соед.; пища, топливо, одежда, лекарства, краски, моющие ср-ва, ВВ, материалы, без к-рых невозможно создание транспорта, книгопечатания, проникновение в космос и проч.,-все это состоит из орг. соединений. Важнейшую роль орг. соед. играют в процессах жизнедеятельности. На стыке органической химии с неорг. химией и биохимией возникли химия металлоорг. соед. и биоорг. химия соотв., широко использующие методы и представления органической химии. Отдельный раздел органической химии составляет химия высокомол. соед.: по величине молекул орг. в-ва делятся на низкомолекулярные (с мол. массой от неск. десятков до неск. сотен, редко до тысячи) и высокомолекулярные (макромолекулярные; с мол. массой порядка 104-106 и более).
Осн. методом органической химии является синтез. Развитие методов синтеза в первую очередь способствовало установлению строения самых сложных соединений. Идеальным завершением процесса определения структуры молекул орг. соед. является полный синтез (тотальный синтез), т.е. получение с помощью совершенно однозначных хим. методов соединения, структура к-рого была предложена на основании изучения др. методами. Орг. синтез-очень тонкое искусство, и химику, приступающему к синтезу, необходимо совершенное сочетание теоретич. и практич. знаний с интуитивным подбором ср-в, наиб. подходящих для построения самых сложных молекул (см. также Органический синтез).
Органическая химия изучает не только соед., получаемые из растит. и животных организмов (т. наз. прир. в-ва), но в осн. соед., созданные искусственно с помощью лаб. или пром. орг. синтеза. Более того, объектами изучения компьютерной органической химии являются соед., не только не существующие в живых организмах, но к-рые, по-видимому, нельзя получить искусственно (напр., гипотетич. аналог метана, имеющий не прир. тетраэдрич. строение, а форму плоского квадрата, в центре к-рого лежит атом С, а в вершинах-атомы Н).
Орг. синтез связывает органическую химию с хим. пром-стью, как малотоннажной (тонкий органический синтез; произ-во лекарств, витаминов, жидких кристаллов, ферментов, феромонов и др.), так и крупнотоннажной (основной органический синтез; произ-во искусств. волокна, пластмасс; переработка нефти и газа и др.).
Строение орг. соед. устанавливают с помощью методов анализа орг. соед., включающих помимо элементного анализа такие физ. методы, как ЯМР, масс-спектрометрия, ИК спектроскопия, рентгеновский структурный анализ, электронография и др.; развиваются также методы выделения, очистки и разделения орг. в-в, напр. разл. виды хрома-тографии.
Классификация органических соединений. Основу орг. соед. составляет незамкнутая (открытая) или замкнутая цепь углеродных атомов; одно или неск. звеньев цепи м. б. заменено на атомы, отличные от углерода,-т. наз. гетероатомы, чаще всего О, N, S. По структуре орг. соед. подразделяют на алифатические соединения - углеводороды и их производные, имеющие открытую углеродную цепь; карбоциклич. соед. с замкнутой углеродной цепью (см. Алициклические соединения, Ароматические соединения); гетероциклические соединения. Углеводороды и их производные, не содержащие кратных связей, относятся к насыщ. соед., с кратными связями - к ненасыщенным.
От каждого углеводорода путем замены атомов Н на разл. функц. группы м. б. образован т. наз. генетич. ряд, напр. этан - этилхлорид - этанол - ацстальдегид - уксусная к-та. В зависимости от типа функц. группы орг. сосд. разделяются на классы: углеводороды RH (функц. группа отсутствует), их галогензамещенные RHal, спирты ROH, альдегиды RCHO, кетоны R2CO, карбоновые к-ты RCOOH, первичные, вторичные и третичные амины RNH2, R2NH и R3N, нитросоед. RNO2; тиолы (меркаптаны) RSH, сульфиды R2S и др. К функц. группам относят также кратные углерод-углеродные связи. Группы орг. соед. однотипной структуры с одинаковыми функц. группами, отличающимися друг от друга по кол-ву групп СН2 в углеродной цепи, составляют гомологический ряд.
Соед., в молекулах к-рых кроме атомов С и Н и атомов-органогенов (Hal, О, N, S) содержатся атомы др. элементов, образующих связи с углеродом, относятся к элементоорга-ническим соединениям (см., напр., Металлоорганические соединения, Мышьякорганические соединения, Фосфороргани-ческие соединения). О правилах наименования орг. соед. см. в ст. Номенклатура химическая.
Историческая справка. Истоки органической химии восходят к глубокой древности (уже тогда знали о спиртовом и уксуснокислом брожении, крашении индиго и ализарином). Однако в средние века (период алхимии) были известны лишь немногие индивидуальные орг. в-ва. Все исследования этого периода сводились гл. обр. к операциям, при помощи к-рых, как тогда думали, одни простые в-ва можно превратить в другие. Начиная с 16 в. (период ятрохимии) исследования были направлены в осн. на выделение и использование разл. лек. в-в: был выделен из растений ряд эфирных масел, приготовлен диэтиловый эфир, сухой перегонкой древесины получены древесный (метиловый) спирт и уксусная к-та, из винного камня-винная к-та, перегонкой свинцового сахара-уксусная к-та, перегонкой янтаря - янтарная. Большая роль в становлении органической химии принадлежат А. Лавуазье, к-рый разработал основные количеств. методы определения состава хим. соединений.
Слияние химии соединений растит. и животного происхождения в единую хим. науку органической химии осуществил Й. Берцелиус, к-рый ввел сам термин и понятие орг. в-ва, образование последнего, по Берцелиусу, возможно только в живом организме при наличии "жизненной силы". Это заблуждение опровергли Ф. Вёлер (1828), к-рый получил мочевину (орг. в-во) из цианата аммония (неорг. в-во), А. Кольбе, синтезировавший уксусную к-ту, М. Бертло, получивший метан из H2S и CS2, A. M. Бутлеров, синтезировавший сахаристые в-ва из формалина. В первой пол. 19 в. был накоплен обширный опытный материал и сделаны первые обобщения, определившие бурное развитие органической химии: развиты методы анализа орг. соед. (Берцелиус, Ю. Либих, Ж. Дюма, М. Шеврёль), создана теория радикалов (Вёлер, Ж. Гей-Люссак, Либих, Дюма) как групп атомов, переходящих неизменными из исходной молекулы в конечную в процессе р-ции; теория типов (Ш. Жерар, 1853), в к-рой орг. соед. конструировались из неорг. в-в-"типов" (тип водорода, воды, хлористого водорода, аммиака) замещением в них атомов на орг. фрагменты; введено понятие изомерии (Берцелиус).
Исследования Э. Франклендом (1852) металлоорг. соед. позволили установить четырехвалентность углерода, заложить основы теории валентности (Ф. Кекуле, 1858) и постулировать существование углерод-углеродных простых и двойных связей. Революц. вклад внес А. Купер (1858), к-рый ввел понятие валентного штриха. С тех пор и по настоящее время химики используют язык т. наз. конституционных (структурных) ф-л молекул орг. соед., в к-рых связи между отдельными атомами обозначаются с помощью одного (простая, или одинарная, связь), двух (двойная) или трех (тройная) валентных штрихов.
Одновременно продолжается интенсивное развитие синтеза. Создаются первые пром. произ-ва орг. соед. (А. Гофман, У. Перкин-старший-синтетич. красители: мовеин, фуксин, цианиновые и азокрасители). Усовершенствование открытого Н. Н. Зининым (1842) способа синтеза анилина послужило основой создания анилинокрасочной пром-сти. В лаборатории А. Байера синтезированы прир. красители - индиго, ализарин, индигоидные, ксантеновые, антрахиноновые.
Идея неразрывной связи хим. и физ. св-в молекулы с ее строением, идея единственности этого строения впервые была высказана Бутлеровым (1861), к-рый создал классич. теорию хим. строения (атомы в молекулах соединяются согласно их валентностям, хим. и физ. св-ва соед. определяются природой и числом входящих в их состав атомов, а также типом связей и взаимным влиянием непосредственно несвязанных атомов). Теория хим. строения определила дальнейшее бурное развитие органической химии: в 1865 Кекуле предложил ф-лу бензола, позднее высказал идею об осцилляции связей; В. В. Марковников и А.М.Зайцев сформулировали ряд правил, впервые связавших направление хим. р-ции с хим. строением вступающего в р-цию в-ва. Эксперим. данные И. Вислиценуса (1873) об идентичности структурных ф-л (+)-молочной к-ты (из кислого молока) и (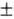 )-молочной к-ты послужили толчком для создания стереохим. теории (Я. Вант-Гофф и Ж. Ле Бель, 1874), в к-рой постулировалось тетраэдрич. строение фрагмента с четырехвалентным атомом углерода, что в случае четырех разл. заместителей предсказывало существование пространственно-зеркальных изомеров; для соед. с двойной связью (тетраэдры соединяются по ребру) - наличие геом. изомерии. На этой основе возникла стереохимия- наука о трехмерной ориентации атомов в молекулах и вытекающих отсюда следствиях, касающихся св-в соед. (см. также Конфигурация стереохими-ческая, Конформационный анализ, Молекулярная механика, Оптическая активность, Хиральностъ).
)-молочной к-ты послужили толчком для создания стереохим. теории (Я. Вант-Гофф и Ж. Ле Бель, 1874), в к-рой постулировалось тетраэдрич. строение фрагмента с четырехвалентным атомом углерода, что в случае четырех разл. заместителей предсказывало существование пространственно-зеркальных изомеров; для соед. с двойной связью (тетраэдры соединяются по ребру) - наличие геом. изомерии. На этой основе возникла стереохимия- наука о трехмерной ориентации атомов в молекулах и вытекающих отсюда следствиях, касающихся св-в соед. (см. также Конфигурация стереохими-ческая, Конформационный анализ, Молекулярная механика, Оптическая активность, Хиральностъ).
Работами Байера, К. Лаара, Л. Клайзена, Л. Кнорра развиты представления о таутомерии - подвижной изомерии. Все эти теоретич. представления способствовали мощному развитию .синтетич. химии. К кон. 19 в. были получены все важнейшие представители углеводородов, спиртов, альдегидов и кетонов, карбоновых к-т, галогено- и нитропроизвод-ных, азот- и серосодержащих структур, гетероциклов арома-тич. природы. Разработаны методы получения диенов, ацетиленов и алленов (А. Е. Фаворский). Открыты многочисл. р-ции конденсации (Ш. Вюрц, А. П. Бородин, У. Перкин, Клайзен, А. Михаэль, Ш. Фридель, Дж. Крафтс, Э. Кнёве-нагель и др.). Исключит. успехи были достигнуты Э. Г. Фишером в изучении углеводов, белков и пуринов, в использовании ферментов в орг. синтезе (1894), им же был осуществлен синтез полипептидов. Основой пром-сти душистых в-в становятся работы О. Валлаха по химии терпенов. Выдающимися даже для нашего времени являются пионерские работы Р. Вильштеттера [установление структуры кокаина (1897) и хлорофилла (1907-11)]. Фундам. вклад в развитие орг. синтеза был внесен В. Гриньяром (1900-20) и Н.Д. Зелинским (1910)-создание исключительно плодотворного метода синтеза магнийорг. соед. и открытие каталитич. превращений углеводородов; последнее сыграло выдающуюся роль в развитии химии нефти. Химия своб. радикалов началась с работ М. Гомберга (1900), открывшим три-фенилметильный радикал, и была продолжена работами А. Е. Чичибабина, Г. Виланда и Ш. Гольдшмидта.
Разработка Ф. Преглем в нач. 20 в. методов микроанализа орг. в-в способствовала дальнейшему быстрому развитию химии прир. соед., что ознаменовалось работами Виланда (1910) по установлению природы желчных к-т, А. Виндауса (1913-15)-природы холестерина, работами Г. Фишера (1927-29) по синтезу таких ключевых соед., как порфирин, билирубин и гемин, У. Хоуорса (Хеуорс)-по установлению структуры углеводов, синтезу витамина С, П. Каррера, Р. Куна (1911-39)-по получению каротиноидов и витаминов В2, В6, Е и К; химия алкалоидов, половых гормонов, терпенов была создана работами А. Бутенандта (1929-61), Л. Ружички (1920-24), А. П. Орехова и Р. Робинсона.
К сер. 20 в. орг. синтез претерпевает бурное развитие. Это определялось открытием таких основополагающих процессов, как получение олефинов с использованием илидов (Г. Виттиг, 1954), диеновый синтез (О. Дильс, К. Альдер, 1928), гидроборирование непредельных соед. (Г. Браун, 1959), синтез нуклеотидов и синтез гена (А. Тодд, X. Корана). Не менее значительны успехи в химии металлоорг. соед. (А. Н. Несмеянов, Г. А. Разуваев). В 1951 был осуществлен синтез ферроцена, установление "сэндвичевой" структуры к-рого Р. Вудвордом и Дж. Уилкинсоном положило начало химии металлоценовых соед. и вообще химии орг. соед. переходных металлов. В 1955 Э.О. Фишер синтезировал дибензолхром и разработал метод синтеза ареновых производных переходных металлов.
В 20-30-е гг. А. Е. Арбузов создает основы химии фос-форорг. соед., что впоследствии привело к открытию новых типов физиологически активных соед., комплексонов и др.
В 60-е гг. Г. Шилл осуществил синтез таких "неклассических" соед., как катенаны и ротаксаны. В 60-80-е гг. Ч. Педерсен, Д. Крам и Ж. М. Лен разрабатывают химию краун-эфиров, криптандов и др. родств. структур, способных образовывать прочные мол. комплексы, и тем самым подходят к важнейшей проблеме "мол. узнавания".
Строение органических соединений. Для орг. соед. характерны неполярные ковалентные связи С—С и полярные ковалентные связи С—О, С—N, С—Hal, С—металл и т.д. Образование ковалентных связей было объяснено на основании развитых Г. Льюисом и В. Косселем (1916) предположений о важной роли электронных образований-октетов и дублетов. Молекула устойчива, если валентная оболочка таких элементов, как С, N, О, Hal, содержит 8 электронов (правило октета), а валентная оболочка водорода-2 электрона. Хим. связь образуется обобществленной парой электронов разл. атомов (простая связь). Двойные и тройные связи образуются соотв. двумя и тремя такими парами. Электроотрицат. атомы (F, О, N) используют для связи с углеродом не все свои валентные электроны; "неиспользованные" электроны образуют неподеленные (свободные) электронные пары. Полярность и поляризуемость ковалентных связей в орг. соед. в электронной теории Льюиса - Кос-селя объясняется смещением электронных пар от менее электроотрицательного к более электроотрицат. атому, что находит выражение в индуктивном эффекте и мезомерном эффекте.
Признание ключевой роли электронных пар сыграло важную роль в классификации орг. соед., к-рые в случае реагентов с четным числом валентных электронов были разделены на нуклеофильные и электрофильные, а р-ции частиц с нечетным числом валентных электронов назвали радикальными.
Классич. теория хим. строения и первонач. электронные представления оказались не в состоянии удовлетворительно описать на языке структурных ф-л строение мн. соед., напр. ароматических. Совр. теория связи в орг. соед. основана гл. обр. на понятии орбиталей и использует молекулярных орбиталей методы. Интенсивно развиваются квантовохим. методы, объективность к-рых определяется тем, что в их основе лежит аппарат квантовой механики, единственно пригодный для изучения явлений микромира. Методы мол. орбиталей в органической химии развивались от простого метода Хюккеля к валентных связей методу, ЛКАО-приближению и др. Широко используются представления о гибридизации атомных орбиталей. Этап проникновения орбитальных концепций в органическую химию открыла резонанса теория Л. Полинга (1931-33) и далее работы К. Фукуи, Вудворда и Р. Хофмана о роли граничных орбиталей в определении направления хим. р-ции. Теория резонанса до сих пор широко используется в органической химии как метод описания строения одной молекулы набором канонич. структур с одинаковым положением ядер, но с разным распределением электронов.
Общая характеристика реакций органических соединений. Р-ции орг. соед. имеют нек-рые специфич. особенности. В р-циях неорг. соед. обычно участвуют ионы; эти р-ции протекают очень быстро, иногда мгновенно при нормальной т-ре. В р-циях орг. соед. обычно участвуют молекулы; при этом одни ковалентные связи разрываются, а другие образуются. Такие р-ции протекают медленнее ионных (напр., десятки часов), и для их ускорения часто требуется повысить т-ру или добавить катализатор. Наиб. часто используют в качестве катализаторов к-ты и основания. Обычно протекает не одна, а неск. р-ций, так что выход нужного продукта очень часто составляет менее 50%. В связи с этим в органической химии употребляют не хим. ур-ния, а схемы р-ций без указания стехиометрич. соотношений.
Р-ции орг. соед. могут протекать очень сложным образом и вовсе не обязательно соответствовать простейшей относит. записи. Как правило, простая стехиометрич. р-ция на самом деле происходит в неск. последоват. стадий. В качестве промежут. соед. (интермедиатов) в многостадийных процессах могут возникать карбкатионы R+, карбанионы R-, своб. радикалы 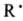 , карбены: СХ2, катион-радикалы (напр.,
, карбены: СХ2, катион-радикалы (напр., 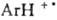 ), анион-радикалы (напр., Аг
), анион-радикалы (напр., Аг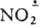 ) и др. нестабильные частицы, живущие доли секунды. Подробное описание всех изменений, к-рые происходят на мол. уровне в процессе превращ. реагентов в продукты, наз. механизмом р-ции.
) и др. нестабильные частицы, живущие доли секунды. Подробное описание всех изменений, к-рые происходят на мол. уровне в процессе превращ. реагентов в продукты, наз. механизмом р-ции.
Исследование влияния строения орг. соед. на механизм их р-ций изучает физическая органическая химия, основы к-рой заложили К. Инголд, Робинсон и Л. Гаммет (1930-е гг.).
Р-ции орг. соед. м. б. классифицированы в зависимости от способа разрыва и образования связей, метода возбуждения р-ции, ее молекулярности и др. (см. Реакции химические).
Взаимод. между реагирующими молекулами с использованием представлений о мол. орбиталях описывается примерно так же, как взаимод. между атомами при образовании молекул. Широкое распространение для этой цели получил метод возмущений мол. орбиталей, на основе к-рого можно предсказать направление (региохимию) и стереохим. результат р-ции, а также саму возможность ее осуществления в данных условиях. Использование граничных орбиталей теории (К. Фукуи, 1952) послужило мощным стимулом к сближению экспериментальной органической химии с квантовой химией. Подлинным триумфом применения метода мол. орбиталей в органической химии явилось опубликование в 1965 правил Вудворда-Хофмана, на основе к-рых можно легко предсказать направление перициклич. р-ций и условия их проведения, необходимые для получения желаемого стереохим. результата (см. Вудворда-Хофмана правила, Перициклические реакции).
Развитие органической химии в настоящее время достигло уровня, позволяющего начать решение такой основополагающей проблемы органической химии, как проблема количеств. соотношения структуры в-ва и его св-ва, в качестве к-рого может выступать любое физ. св-во (напр., т-ра плавления), биол. активность любого строго заданного типа (напр., пестицидная) и др. Решение задач такого типа осуществляется с использованием мат. методов.
Возникновение органических соединений. Большинство орг. соед. в природе образуется в процессе фотосинтеза из диоксида углерода и воды под действием солнечного излучения, поглощаемого хлорофиллом в зеленых растениях. Однако орг. соед. должны были существовать на земле и до возникновения жизни, к-рая не могла появиться без них. Первичная земная атмосфера около 2 млрд. лет назад имела восстановит. св-ва, т. к. в ней не было кислорода, а содержались прежде всего водород и вода, а также СО, азот, аммиак и метан.
В условиях сильного радиоактивного излучения земных минералов и интенсивных атм. разрядов в атмосфере протекал абиотич. синтез аминокислот по схеме:
CH4 + H2O + NH3 Аминокислоты
Аминокислоты
Возможность такой р-ции в настоящее время доказана лаб. опытами. Аминокислоты (из к-рых состоят белки) накапливались в океане вместе с др. в-вами и постепенно превращались во все более сложные орг. в-ва, пока, наконец, не появилась возможность создания живой клетки.
Лит.: Чичибабин А. Е., Основные начала органической химии, 6 изд., т. 1-2, М., 1954-58; Каррер П., Курс органической химии, пер. с нем., 2 над., Л., 1962, Ингольд К., Теоретические основы органической химии, 2 изд., пер. с англ., М., 1973; Быков Г. В., История органической химии. Структурная теория, физическая органическая химия, расчетные методы, М., 1976; Дьюар М., Догерти Р., Теория возмущений молекулярных орбиталей в органической химии, пер. с англ., М., 1977; Быков Г. В., История органической химии. Открытие важнейших органических соединении. М., 1978; Общая органическая химия, под ред. Д. Бартона и У. Д. Оллнса, пер. с англ., т. 1-12, М., 1981-88; Терней А., Современная органическая химия, пер. с англ., т. 1-12, М., 1981; М а рч Д., Органическая химия. Реакции, механизмы и структура, пер. с англ., т. 1-4. М., 1987-88; Beilsteins Handbuch der organischen Chemie, 4 Aufl., bearb. von B. Prager [u.a.], Bd 1-31, В., 1918-40 (с 1928 г. изд. доп. тт.); Houben -Weyl, Methoden der organischen Chemie, 4 Aufl., Bd 1 20, Stuttg., 1952-88.
Я. С. Зефиров.