Спектроскопические методы исследования органических веществ
Возможность изучения размеров и формы отдельных молекул и относительного расположения в них атомов и радикалов, и особенно результаты рентгеноструктурных исследований, дали новый толчок изучению физических свойств веществ с целью более глубокого проникновения в детали структуры молекул. Особенно интересны исследования электрических и магнитных свойств кристаллов, в частности оптической, электрической и магнитной анизотропии, в зависимости от тонкой структуры кристалла.
В настоящее время для исследования строения органических веществ широ,ко применяется изучение их инфракрасных, видимых и ультрафиолетовых спектров поглощения. Инфракрасные и комбинационные спектры связаны с колебательными и вращательными движениями атомов (точнее, ядер атомов), видимые и ультрафиолетовые спектры обязаны своим происхождением электронным переходам.
Молекулярные спектры имеют значительно более сложную структуру по сравнению с атомными спектрами. Эта сложность молекулярных спектров обусловлена тем, что в процессах, связанных с энергетическими переходами в молекуле наряду с электронами участвуют и ядра, движение которых и находит свое отображение в молекулярных спектрах. Ядра атомов в молекуле могут совершать два рода движений: вращательное движение вокруг центра тяжести молекулы и колебательное движение около некоторых положений равновесия. Оба рода движений являются квантованными, что, в частности, проявляется в дискретной структуре молекулярных спектров.
Молекулярные спектры можно разделить на три класса: вращательные спектры, связанные с вращением ядер в молекуле, колебательные спектры, связанные с колебанием ядер, и электронные спектры, связанные с движением электронов (электронные переходы). Первые два рода спектров лежат в инфракрасной области.
Если изучение атомных спектров дало ряд ценнейших сведем ний для создания теории атома, то изучение молекулярных спектров играет очень важную роль при исследовании строения молекул. При помощи спектроскопических исследований можно найти межатомные расстояния в молекулах, собственные частоты колебаний ядер и др. Эти данные вместе с дипольными моментами, а также с данными рентгенографического и электронографического анализа дают возможность составить надежное детальное представление о строении молекул. Спектроскопическими методами можно определить также энергию диссоциации молекул. Пользуясь молекулярным спектральным анализом, можно производить идентификацию химических соединений и измерять их концентрации.
Так как отдельным радикалам (например, ОН, NH2, NO2, СО, С6Н5 и т. д.), а также отдельным связям внутри молекулы, (например, С=С, С≡С, С=О, С—Н и т. д.) соответствуют определенные характеристические частоты в инфракрасных, спектрах и спектрах комбинационного рассеяния (мало изменяющиеся от соединения к соединению), то по этим спектрам можно судить о наличии в молекуле тех или иных радикалов или связей.
Эффект комбинационного рассеяния, одновременно открытый в 1928 г. советскими физиками Г. С. Ландсбергом и Л. О. Мандельштамом и индийским ученым Ч. В. Раманом, заключается в том, что при освещении жидкости сильным источником монохроматического света (например, мощной ртутной лампой со светофильтром, пропускающим фиолетовую линию 4047 Å) в спектре рассеянного света наряду с линией, имеющей частоту ν0, падающего света, наблюдаются слабые линии — спутники, смещенные на равную величину в обе стороны, с частотами ν0—ν' и, ν0 + ν', ν0—ν" и ν0 + ν", ν0—ν'" и ν0 + ν'" и т. д. Эти симметричные спутники, однако, отличаются по своей интенсивности: интенсивности линий с частотами, большими, чем ν0, значительно слабее, и наблюдение их очень затруднительно. Поэтому в, основном говорят обычно о системе спутников ν0—ν' ν0—ν", ν0—ν" и т. д. Оказывается, что величины смещения частот (ν', ν", ν'" . . .) отвечают переходам данной молекулы от одного колебательного уровня к другому, т. е. отвечают собственным колебаниям, возникающим в молекуле. Эти величины смещения не зависят от частоты ν0 падающего света.
Для сложных органических молекул расшифровать расчетным путем наблюдаемые частоты и отнести их к колебаниям тех или иных связей и группировок в молекуле практически невозможно. Однако возможен иной путь. Изучают и сравнивают спектры ряда гомологов или аналогов и стремятся выявить характеристические частоты для определенных классов соединений.
На диаграмме (рис. 54) сопоставлены спектры комбинационного рассеяния ряда галоидпроизводных парафиновых углеводородов. Высота отдельных штрихов, изображающих положение наблюдаемых в спектрах линий, соответствует относительной интенсивности этих линий. По оси абсцисс отложены частоты колебаний для этих линий, выраженные в так называемых обратных сантиметрах (см–1). Сравнивая спектры для СН4 и ССl4
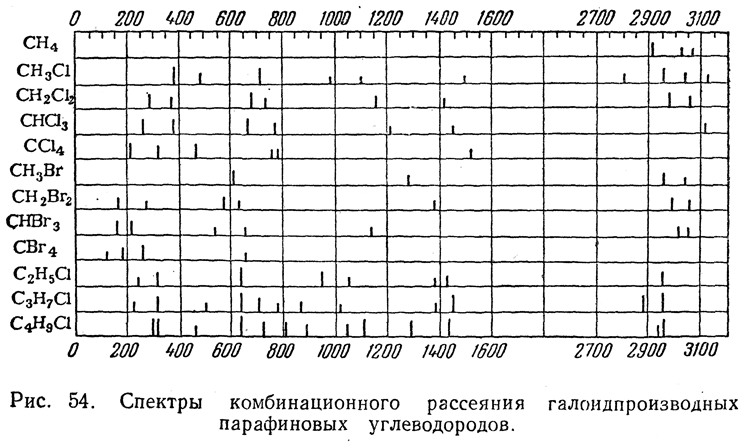
или СВr4, мы видим прежде всего, что у полностью замещенных метанов отсутствует группа линий, лежащих между 2900 и 3150 см–1, а у СН4 отсутствуют группы линий между 210 и 800 см–1 или соответственно между 150—700 см–1. Отсюда можно сделать вывод, что группа линий 2900—3150 см–1 отвечает связям С—Н, а соответствующие группы линий, лежащие у красного конца спектра, — связям С—Сl или С—Вr. Группы слабых линий около 1000—1500 см–1 в спектрах" хлорпроизводных или 1150—1450 см–1 в спектрах бромпроизводных, очевидно, также обязаны своим происхождением наличию в молекуле соответствующих галоидов.
Для некоторых функциональных групп можно приурочить в спектрах комбинационного рассеяния узко определенный, характерный для присутствия этих групп в молекуле интервал частот. Об этих характеристических частотах дают представление, например, данные, приведенные в табл. 32.
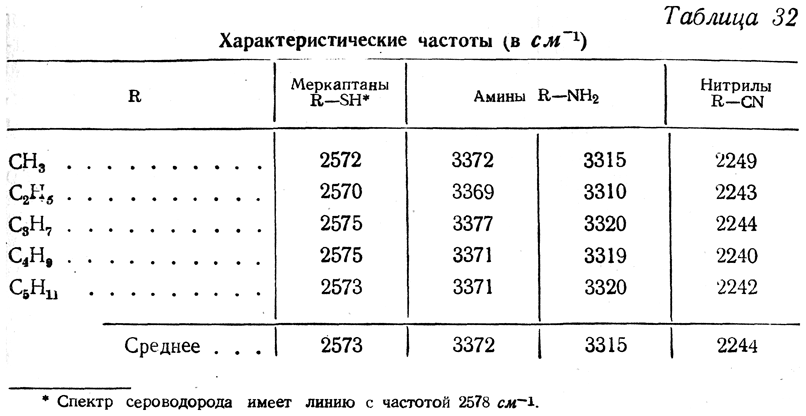
Частоты колебаний, превышающие 2500 см–1, характерны для связей с водородом — самым легким элементом. Частоты валентных колебаний (т. е. смещений вдоль линии, соединяющей ядра связываемых атомов) для связей С—Н лежат между 2800 и 3300 см–1, для О—Н — между 3350 и 3450 см–1 и для N—Н составляют ~3300 см–1.
Частоты ординарных связей С—С лежат между 800 и 1200 см–1. Однако в тот же интервал попадают частоты связей С—О 1032 см–1 для СН3ОН), С—N (1037 см–1 для CH3—NH2), С—F (1040 см–1 для СН3—F). Отличить и приурочить линии с частотами в этой области (800—1200 см–1) для более сложных органических соединений оказывается затруднительным.
Частоты двойной связи С=С в олефинах колеблются к пределах 1620—1680 см–1 в зависимости от характера замещения; связь С—О в кетонах сохраняет частоту ~1710 см–1, в альдегидах ~1720 см–1, в эфирах карбоновых кислот ~1735 см–1. Но у самих карбоновых кислот она оказывается значительно пониженной и составляет ~1650 см–1.
Тройная связь в ацетилене обладает частотой 1960 см–1; у моноалкилацетиленов она повышается до 2120 см–1, а у двузамещенных— до 2234 см–1. Тройная связь C≡N обладает частотой ~2250 см–1.
С помощью спектров комбинационного рассеяния можно решать такие сложные задачи, как установление конфигурации цис- и транс-изомеров, определение числа устойчивых конформаций, фактически существующих при данной температуре, примерная оценка величины эффекта сопряжения простых связей с кратными и пр.
Спектры комбинационного рассеяния широко применяются не только для чисто теоретических исследований в области органической химии, но и для целей идентификации и анализа органических соединений. Группой советских ученых (Г. С. Ландсберг, Б. А. Казанский и др.) разработан метод анализа этим путем состава бензиновых фракций нефти.
Очень многие из задач, о которых выше шла речь, можно решать также при помощи инфракрасных спектров.
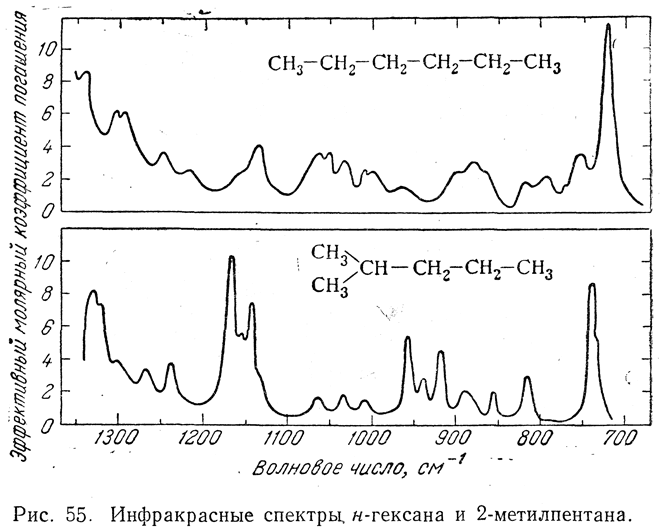
На рис. 55 видно, насколько различаются инфракрасные спектры веществ при сравнительно небольшом изменении их строения. В инфракрасных спектрах, как и в спектрах комбинационного рассеяния, отдельным радикалам и связям отвечают определенные характеристические частоты, что часто позволяет выбрать для впервые полученного соединения наиболее правдоподобное строение. Кроме того, для получения инфракрасных спектров требуется меньше вещества и времени, чем для снятия спектров комбинационного рассеяния. Поэтому некоторые задачи установления строения веществ и качественного анализа часто проще решать методом инфракрасных спектров. Зато количественный анализ в большинстве случаев легче и тоньше производится при помощи спектров комбинационного рассеяния. Кроме того, многие характерные линии отдельных группировок и связей проявляются либо только в инфракрасных спект-
pax, либо в спектрах комбинационного рассеяния. Таким образом, эти два метода взаимно дополняют друг друга.
Более частое применение методов инфракрасной спектроскопии зарубежными химиками объясняется лишь тем, что в их странах не налажено производство достаточно совершенных спектрографов для проведения точных исследований с помощью спектров комбинационного рассеяния.
Спектры поглощения в видимой и ультрафиолетовой области также позволяют решать задачи, названные выше. Однако поглощением в этой области спектра обладают не все вещества, а главным образом соединения ароматического характера и соединения, содержащие в молекуле большое число двойных связей.