Абсолютная конфигурация, стерические ряды и вальденовское обращение
При рассмотрении пространственного строения веществ с асимметрическими атомами различают их относительную и абсолютную конфигурации. Относительная конфигурация — это взаимное расположение заместителей при разных асимметрических атомах по отношению друг к другу; обычно ее обозначают приставками к основному названию вещества (цис- и транс-, трео- и эритро-, мезо-, алло- и др.). Абсолютная конфигурация — это истинное расположение в пространстве заместителей при каждом асимметрическом атоме молекулы; чаще всего ее обозначают буквами D или L. Например, правовращающая и левовращающая винные кислоты обладают противоположными (антиподными) абсолютными конфигурациями, но одинаковой относительной конфигурацией, отличающейся в то же время от относительной конфигурации мезовинной кислоты
(различные расстояния между группами ОН, СООН и т. д.):
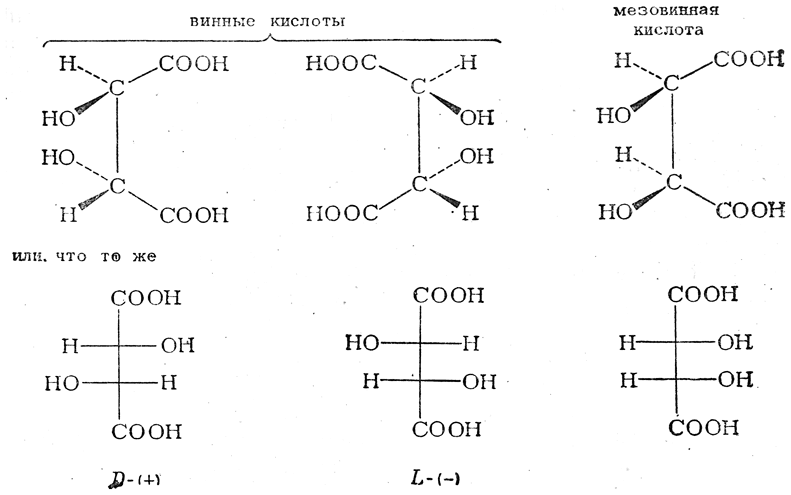
Относительная конфигурация многих веществ может быть выяснена различными химическими и физическими методами. Прямое же определение абсолютной конфигурации, напротив, представляет очень трудную задачу, которую пока удалось решить только на одном примере: в 1951 г. путем рентгеноструктурного анализа калий-рубидиевой соли D(+)-винной кислоты было установлено, что эта кислота обладает абсолютной конфигурацией, изображаемой приведенными выше формулами. Тем не менее одного этого эксперимента достаточно, чтобы решить вопрос об истинном пространственном строении огромного числа оптически деятельных соединений путем корреляции (установления соответствия) их абсолютных конфигураций и сведения этих соединений в стерические ряды.
В качестве стереохимического стандарта еще в 1906 г., по предложению М. А. Розанова, был избран глицериновый альдегид, (+)-форме которого была произвольно приписана формула (I), а (—)-форме — формула (II):

С тех пор абсолютные конфигурации других оптически деятельных соединений рассматривались как соответствующие либо правовращающему, либо левовращающему антиподу глицеринового альдегида, в результате чего возникли два ряда конфигуративно родственных соединений — D-ряд и L-ряд.
При этом корреляция конфигураций различных веществ чаще всего осуществлялась химическим путем. Например, (+)-глицериновый альдегид был окислен в (—)-глицериновую кислоту, которая была получена также из (+)-изосерина при его диазотировании; (+)-изосерин в свою очередь был дезаминирован с образованием (—)-молочной кислоты и получен из (+)-винной кислоты через (—)-хлоряблочную и (+)-яблочную кислоты:
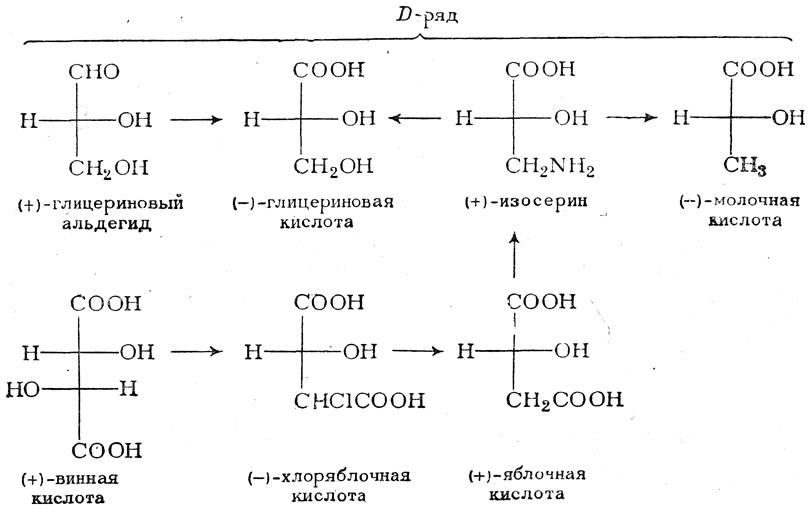
Отсюда следовало, что все эти соединения обладают одинаковой конфигурацией, которая была обозначена как D-конфигурация по основному члену ряда — D(+)-глицериновому альдегиду.
Сравнивая выведенную этим путем проекционную формулу (+)-винной кислоты с установленной на основании рентгеноструктурных данных ее пространственной формулой, можно легко убедиться, что сделанный ранее произвольный выбор проекционной формулы (+)-глицеринового альдегида случайно оказался правильным. Следовательно, абсолютные конфигурации теперь уже не являются условными, а правильно отражают истинное пространственное строение молекул.
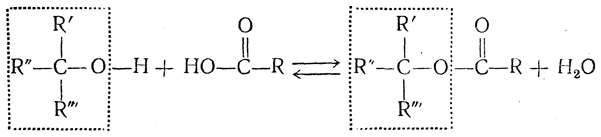
При сведении различных веществ на основании их взаимных превращений в один стерический ряд возникает вопрос, остается ли при химических реакциях конфигурация молекулы неизменной? Оказывается, что при всех превращениях, при которых не происходит разрыв связи у асимметрического атома, конфигурация молекулы всегда сохраняется. Например, при ацилировании спиртов, гидроксил которых присоединен к асимметрическому атому углерода, и при омылении образовавшихся сложных эфиров связь между атомом кислорода и асимметрическим углеродным атомом не разрывается, и потому изменения конфигурации не происходит:
Если же замещение происходит непосредственно при асимметрическом центре, то конфигурация молекулы может измениться. Например, из D(+)-α-бромпропионовой кислоты при действии на нее разбавленной щелочи получается D(—)-молочная кислота, а при действии концентрированной щелочи — L(+)-молочная кислота:
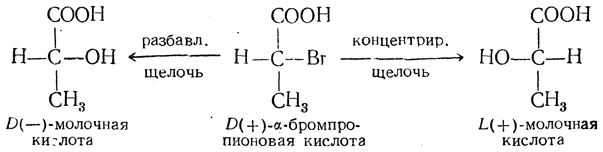
Как видно из приведенной схемы, пространственное расположение заместителей у асимметрического атома при взаимодействии с разбавленной щелочью не изменилось, тогда как в результате реакции с концентрированной щелочью молекула приняла конфигурацию, противоположную исходной. Это уже упоминавшееся ранее явление вальденовского обращения, или инверсии конфигурации, встречается весьма часто и имеет очень большое теоретическое значение. Практически оно может быть использовано для получения некоторых ценных соединений из их более доступных стереоизомеров.
Современную теорию вальденовского обращения целесообразно рассмотреть на примере нуклеофильного замещения, т. е реакции, при которой асимметрический центр подвергается атаке аниона или отрицательного конца диполя полярной молекулы. Такое замещение может протекать либо по бимолекулярному (SN2), либо по мономолекулярному (SN1) механизму, но только бимолекулярное замещение всегда приводит к вальденовскому обращению.
При бимолекулярной реакции по мере приближения аниона X– к молекуле I происходит постепенное удаление радикала Z и одновременное отталкивание трех остальных заместителей, так что в переходном состоянии II эти заместители находятся в одной плоскости. Последующее отщепление аниона Z– и образование связи между центральным атомом и новым заместителем X приводит к конфигурации (III), которая являлась бы зеркальным отражением исходной конфигурации (I), если бы X был тождественен Z:
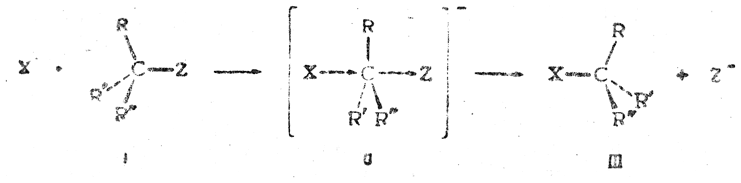
Квантово-механические расчеты показывают, что энергии активации реакции минимальна при атаке входящего заместителя со стороны, противоположной отщепляющемуся заместителю. Поэтому при бимолекулярном нуклеофильном замещении всегда должно происходить вальденовское обращение. Известные до настоящего времени факты подтверждают это положение.
В случае мономолекулярного нуклеофильного замещения первая фаза реакции заключается в диссоциации молекулы (I). Образующийся карбониевый катион (II) принимает плоскую форму (III), и присоединение к нему атакующего аниона X– справа или слева равновероятно. Поэтому в результате реакции получается равное число молекул с противоположными конфигурациями (IV и V), т. е. происходит рацемизация:
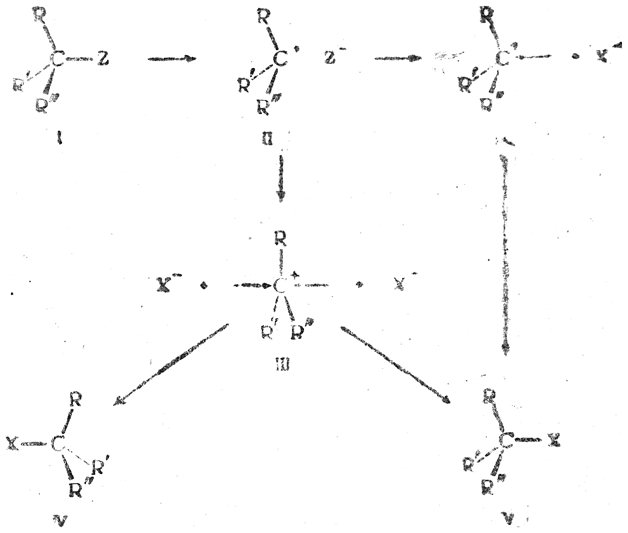
Если же стабильность катиона в пирамидальной форме (II) повышена из-за особенностей его электронного строения (наличие соответствующим образом расположенных ионизированных групп, неподеленных электронных пар, π-электронов кратных связей и т. п.), то переход его в плоскую форму (III) может совершаться медленнее, чем присоединение аниона X–, которое в этом случае приводит к конфигурации (V), аналогичной имевшейся в исходной молекуле (I).
Примерами двух типов нуклеофильного замещения являются рассмотренные на стр. 593 реакции гидролиза D(+)-α-бpoмпропионовой кислоты. При действии на это соединение ионов ОН– (в концентрированной щелочи) происходит обмен брома на гидроксил по механизму SN2 с вальденовским обращением При действии же менее нуклеофильного диполя Н—ОН (в разбавленной щелочи) реакция протекает по механизму SN1. Промежуточный катион типа II стабилизуется при этом в пирамидальной форме вследствие внутримолекулярного взаимодействия С+ с СОО–, и конфигурация сохраняется.
Возможности достоверно предсказывать механизм реакции замещения у асимметрического центра до сих пор не имеется. Часто при действии близких по характеру реагентов, и даже одного и того же реагента в различных условиях, реакция может протекать с сохранением или обращением конфигурации исходного оптически деятельного соединения. Поэтому при установлении конфигурации какого-либо вещества всегда стремятся превратить его в соединения с уже известным пространственным строением путем последовательных превращений, не затрагивающих асимметрического атома.
Однако для определения пространственного строения некоторых веществ этот путь оказывается непригодным ввиду невозможности их превращения в соединения с известной конфигурацией без участия в реакции асимметрического центра молекулы. В таких случаях используют другие методы, из которых наиболее важными в настоящее время являются кинетический метод, асимметрический синтез, спектрополяриметрия и рентгеноструктурный анализ.
Применение кинетического метода для определения конфигурации основано на том, что установление бимолекулярного механизма реакции нуклеофильного замещения считается равносильным доказательству наличия инверсии. С помощью этого метода была, например, определена конфигурация природного (—)-серина. Это соединение, путем замены в нем гидроксильной группы на хлор и последующего восстановления, т. е. путем реакций, не затрагивающих асимметрический центр, превращается в (+)-аланин; оба соединения должны, следовательно, иметь одинаковую конфигурацию. Тот же (+)-аланин получается из (+)-α-бромпропионовой кислоты, которая при щелочном гидролизе дает L-(+)-молочную кислоту:
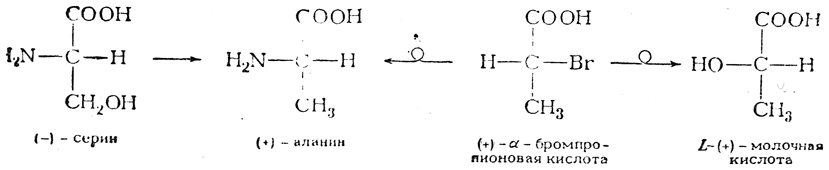
Как показывают кинетические измерения, обе реакции нуклеофильного замещения при асимметрическом центре (+)-α-бромпропионовой кислоты протекают по бимолекулярному механизму и, следовательно, сопровождаются вальденовским обращением (в схеме это показано кружком на стрелке, указывающей направление реакции). Двойная инверсия равноценна сохранению конфигурации, и поэтому (+)-аланин, а следовательно, и (—)-серии должны быть отнесены к тому же L-ряду, что и (+)-молочная кислота, в то время как (+)-α-бромпропионовая кислота относится к D-ряду.
Асимметрический синтез может быть использован, например, для определения абсолютной конфигурации оптически деятельных спиртов, у которых асимметрический атом углерода связан с гидроксилом и тремя существенно различными по величине заместителями (Б — наибольший заместитель, Ср — средний, М — меньший по эффективному объему):

Если при помощи хлорангидрида фенилглиоксиловой кислоты превратить спирт I в кетоэфир III и подействовать на него реактивом Гриньяра, то образуются сложные эфиры L(+)- и D(—)-форм атролактиновой кислоты (IV и V):
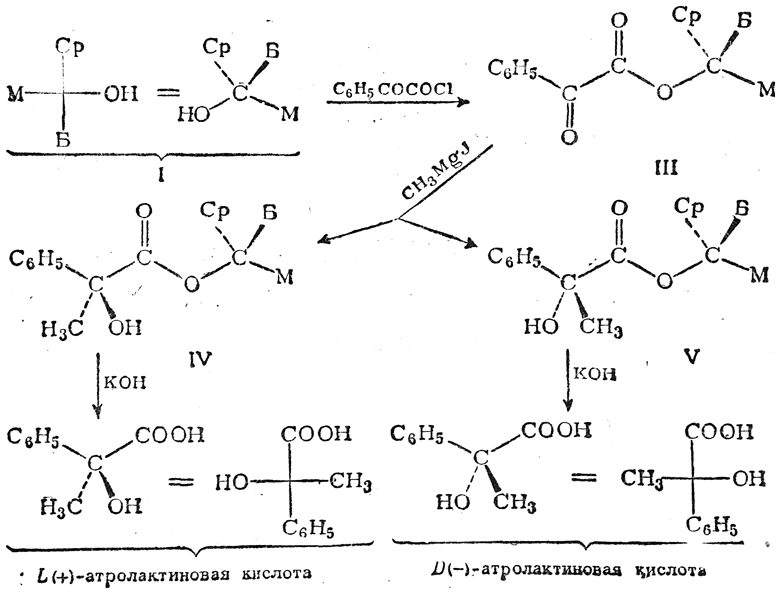
При этом соединение IV получится в несколько большем количестве, чем V, так как присоединение метальной группы к кетонному карбонилу в молекуле кетоэфира III происходит легче со стороны заместителя Ср (из-за плоскости чертежа), чем со стороны большего по размерам заместителя Б. Поэтому после омыления смеси сложных эфиров IV и V получится смесь L(+)- и D(—)-атролактиновых кислот, в которой преобладает L(+)-форма, вследствие чего эта смесь кислот не является рацемической, а вращает плоскость поляризации света вправо. Если бы исходный спирт обладал конфигурацией II, то в результате тех же превращений получилась бы атролактиновая кислота, содержащая избыток молекул D(—)-антипода и потому вращающая влево. Таким образом, определив направление вращения получившейся атролактиновой кислоты, можнo установить пространственное строение исходного спирта. Этот метод позволил выяснить абсолютные конфигурации в ряду терпенов, стероидов и некоторых других сложных природных соединений.
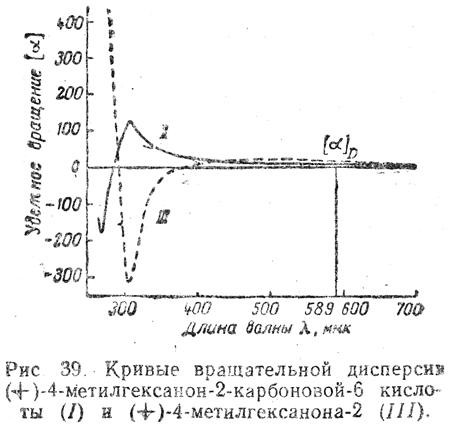
Спектрополяриметрический метод основан на изучении физического свойства, наиболее характерного для каждого асимметричного вещества, а именно его оптической активности. Раньше уже указывалось, что направление вращения плоскости поляризации света само по себе не определяет принадлежности соединения к D- или L-ряду. Если два сходных по структуре вещества имеют не только одинаковые по знаку, но и близкие по величине [α]D, это также не означает, что они обладают одинаковым пространственным строением. Идентичность конфигураций этих веществ может быть установлена лишь путем измерения их оптической активности на протяжении всей видимой и доступной ультрафиолетовой частей спектра и сравнения получающихся при этом кривых зависимости величины вращения от длины волны (так называемых кривых вращательной дисперсии). Например, полученная из природного антнбиотика актидиона (+)-4-метилгексанон-2-карбоновая-6 кислота имеет примерно такое же значение [α]D, как (+)-4-метилгексанон-2, для которого установлена абсолютная конфигурация III:
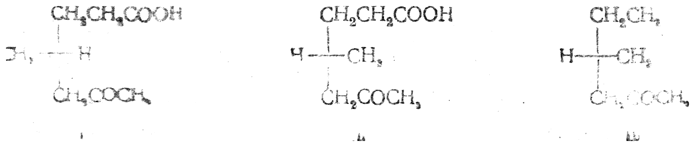
Тем не менее (+)-4-метилгексанон-2-карбоновой-6 кислоте следует приписать формулу I, а не формулу II, так как кривая ее вращательной дисперсии (рис. 39) имеет при ~ 300 ммк пик (положительный эффект Коттона), тогда как кривая вращательной дисперсии 4-метилгексанона-2 имеет при этой же длине волны впадину (отрицательный эффект Коттона).
Спектрополяриметрия особенно удобна для установления абсолютной конфигурации различных алифатических и алициклических карбонильных соединений, но она успешно применяется также и к другим классам органических веществ.
Другим важнейшим методом установления конфигураций является рентгеноструктурный анализ, который позволяет определять пространственное строение самых разнообразных органических молекул.