Лизосомы
А. Структура и состав
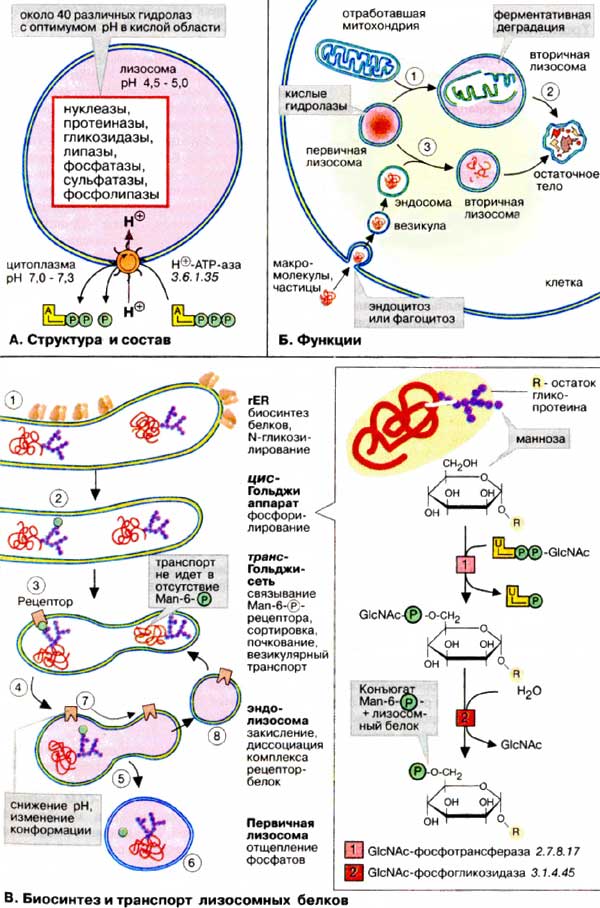
Лизосомы — это органеллы диаметром 0,2-2,0 мкм, окруженные простой мембраной, способные принимать самые разные формы. Обычно на клетку приходится несколько сотен лизосом. Функция лизосом заключается в деградации клеточных компонентов. Деградация достигается за счет присутствия в лизосомах около 40 типов различных расщепляющих ферментов — гидролаз с оптимумом действия в кислой области. Главный фермент лизосом — кислая фосфатаза. В мембране лизосом находятся АТФ-зависимые протонные насосы вакуольного типа. Они обогащают лизосомы протонами, вследствие чего для внутренней среды лизосом рН 4,5-5,0 (в то время как в цитоплазме рН 7,0-7,3). Лизосомные ферменты имеют оптимум рН около 5,0, т. е. в кислой области. При рН, близких к нейтральным, характерным для цитоплазмы, эти ферменты обладают низкой активностью. Очевидно, это служит механизмом защиты клеток от самопереваривания о том случае, если лизосомный фермент случайно попадет в цитоплазму.
Б. Функции
Главная функция лизосом — ферментативная деградация попавших в них макромолекул и органелл. Примером может служить деградация отработавших митохондрий по механизму аутофагии (захвата органеллы) (1). После захвата органеллы первичные лизосомы превращаются во вторичные, в которых и идет процесс гидролитического расщепления (2). В итоге образуются «остаточные тела», состоящие из негидролизовавшихся фрагментов. Лизосомы ответственны также за деградацию макромолекул и частиц, захваченных клетками путем эндоцитоза и фагоцитоза, например липопротеинов, протеогормонов и бактерий (гетерофагия). В этом случае лизосомы сливаются с эндосомами (3), содержащими вещества, подлежащие деградации.
В. Биосинтез и транспорт лизосомных белков
Первичные лизосомы образуются в аппарате Гольджи.
Лизосомные белки синтезируются в ШЭР, где они гликозилируются путем переноса олигосахаридных остатков (1, см. с. 226). На последующей стадии, типичной для лизосомных белков, терминальные маннозные остатки (Man) фосфорилируются no C-6 (на схеме справа). Реакция протекает в две стадии. Сначала на белок переносится GlcNAc-фосфат, а затем идет отщепление GlcNAc. Таким образом, лизосомные белки в процессе сортировки приобретают концевой остаток маннозо-6-фосфата (Man-6-P, 2).
В мембранах аппарата Гольджи имеются молекулы-рецепторы, специфичные для Man-6-P-остатков и за счет этого специфически узнающие и селективно связывающие лизосомные белки (3). Локальное накопление этих белков происходит с помощью клатрина. Этот белок позволяет вырезать и транспортировать подходящие мембранные фрагменты в составе транспортных везикул к эндолизосомам (4), которые затем созревают с образованием первичных лизосом (5) в заключение от Man-6-P отщепляется фосфатная группа (6).
Man-6-P-рецепторы используются вторично в процессе рецикла. Снижение рН а эндолизосомах приводит к диссоциации белков от рецепторов (7). Затем рецепторы с помощью транспортных везикул переносятся обратно в аппарат Гольджи (8).
Некоторые редко встречающиеся заболевания связаны с генетическими дефектами лизосомных ферментов, так как эти ферменты участвуют в деградации гликогена (гликогенозы), липидов (липидозы) и протеогликанов (мукополисахаридозы). Продукты, которые не могут участвовать в метаболизме из-за дефектов или отсутствия соответствующих ферментов, накапливаются в остаточных телах, что приводит к необратимому повреждению клеток и как результат к нарушению функций соответствующих органов.