Электрические свойства молекул и дипольный момент
Для познания природы органических молекул много дали исследования их электрических свойств. Начало этим исследованиям положил Дебай еще в 1912 г.
Исследуя диэлектрические постоянные растворенных веществ при разных температурах, Дебай нашел, что диэлектрические постоянные некоторых веществ не изменяются при изменениях температуры, тогда как для других веществ в растворах и в газообразном состоянии они зависят от температуры. Изменение поляризации с температурой Дебай объяснил тем, что молекулы этих веществ электрически полярны и что поэтому в электрическом поле, кроме обычной поляризации (в результате деформации молекул), происходит также поляризация вследствие определенной ориентации молекул-диполей по отношению к силам электрического поля. Если вещество находится в газообразном или растворенном состоянии, эта ориентация молекул-диполей нарушается вследствие теплового движения молекул. Поэтому слагающая поляризации, зависящая от ориентации молекул-диполей, уменьшается с повышением температуры
Молекулярная поляризация веществ этого рода в газообразном состоянии и в растворах может быть вычислена по формуле Дебая:
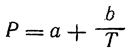
Здесь а — постоянная, а величина b определяется из следующего выражения:
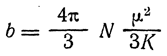
где N — число молекул в грамм-молекуле вещества (число Авогадро);
К — постоянная;
μ — электрический момент молекулы-диполя, который и получил название дипольного момента.
Приведенное уравнение дает возможность вычислить дипольные моменты на основании экспериментальных данных для веществ в газообразном состоянии и в виде растворов в неполярных растворителях (гексан, сероуглерод, бензол и пр.).
Полярность молекул, бесспорно, подтверждается опытами, показавшими отклонение путей молекул, движущихся в электрическом поле.
С точки зрения электронных представлений о природе химической связи полярность молекул объясняется следующим образом.
Ковалентная связь, осуществляемая общей парой электронов, редко оказывается электрически симметричной. Это имеет место только при соединении одинаковых атомов или групп

или атомов с близким электронным сродством.
Если атомы (атомные ядра) или их группы обладают неодинаковым сродством к электронам, симметрия σ-связи, т. е. симметрия расположения электрических зарядов, нарушается; так будет, например, в случаях:

Такое смещение электронов связи часто обозначают стрелкой, направленной в ту сторону, в которую, как предполагается, смещены электроны:
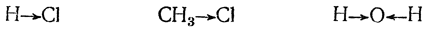
Иногда стрелку ставят посредине ковалентного штриха, например:
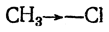
Величину смещения электронов характеризует величина дипольного электрического момента:
μ = e-r
где е — величина элементарного заряда, а r — расстояние между центрами тяжести положительных зарядов ядер и отрицательных зарядов электронных облаков у данной связи или у данной молекулы.
Таким образом, порядок величины дипольного момента определяется произведением элементарного заряда (4,8 ∙ 10–10 эл.-ст. ед.) на длину, которая для межатомных расстояний в молекулах близка к 10–8 см. Поэтому удобно выражать величины дипольных моментов в так называемых единицах Дебая (D), равных 10–10 ∙ 10–8=10–18 эл.-cт. ед.∙см.
В случаях, когда в молекуле полярна лишь одна связь, величина дипольного момента определяет полярность этой связи; в тех же случаях, когда в молекулах имеется несколько полярных связей, величина дипольного момента определяет лишь суммарную величину полярности молекулы. Поэтому величины дипольных моментов молекул, состоящих из двух атомов, представляют особый интерес.
Если электронная пара окончательно переходит в безраздельное обладание одного из партнеров связи, вместо ковалентной связи возникает, как уже раньше отмечалось, ионная связь.
Для чисто ковалентной (гомеополярной) связи дипольный момент должен равняться нулю, а для чисто ионной связи он должен был бы измеряться произведением заряда электрона (4,8 ∙ 10–10 эл.-ст. ед.) на сумму атомных радиусов rA + rB обоих партнеров связи — элементов А и В.
Оказалось, что μ = 0 для следующих молекул:
1. Одноатомные газы: Не, Ne, Аr, Кr, Хе, пары К, Na.
2. Симметричные двухатомные газы типа А—А: Н2, N2, О2, Сl2.
3. Симметричные линейные трехатомные, четырехатомные и т. д. молекулы типа В—(А)n—В: О =С=О, S=C=S,
H—C≡C—H.
4. Симметричные тетраэдрические молекулы типа АВ4: СН4, ССl4, SiCl4, SnJ4.
Существенно отличный от нуля дипольный момент имеют: 1. Несимметричные двухатомные молекулы типа А—В:
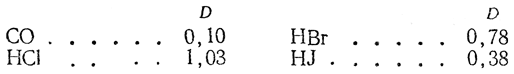
2. Несимметричные линейные молекулы типа В—А—С;
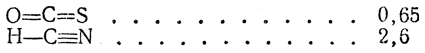
3. Нелинейные молекулы типа В—А—В:
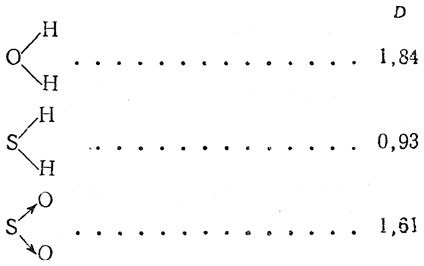
4. Молекулы типа АВ3:
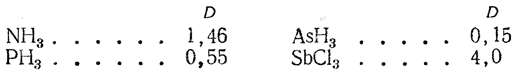
Наличие дипольного момента у таких молекул, как Н2О, H2S, объясняется тем, что связи у кислорода и серы расположены под углом; по квантово-механическим соображениям этот угол должен быть равен 90°, однако он несколько искажается вследствие взаимного отталкивания заместителей. Поэтому у молекулы воды, например, угол НОН оказывается равным ~105°.
Учитывая, что дипольные моменты, как величины направленные, должны подчиняться правилу векториального сложения, мы можем по дипольному моменту воды, зная величину угла НОН, построить параллелограмм моментов, вносимых каждой связью О—Н, и найти их величину. Эта величина μOH оказывается равной 1,51 D.
Молекула аммиака обладает значительным моментом. Для нее была доказана пирамидальная структура, причем плоский угол при вершине пирамиды, где находится ядро азота (угол HNH), составляет ~107°. Расчет, аналогичный приведенному выше, дает для момента связи N—Н величину μNH=1,31 D.
Что касается органических соединений, то здесь оказалось, что не только для СН4 и СН3—СН3, но и вообще для всех предельных углеводородов дипольный момент равен нулю.
В табл. 31 сопоставлены дипольные моменты некоторых органических соединений, обладающих функциональными заместителями. Из данных табл. 31 можно сделать вывод, что величина дипольного момента у производных предельных углеводородов определяется в основном функциональной группой, оставаясь практически почти постоянной (или слабо возрастая) в пределах гомологического ряда (небольшие отклонения наблюдаются лишь у первых членов ряда).
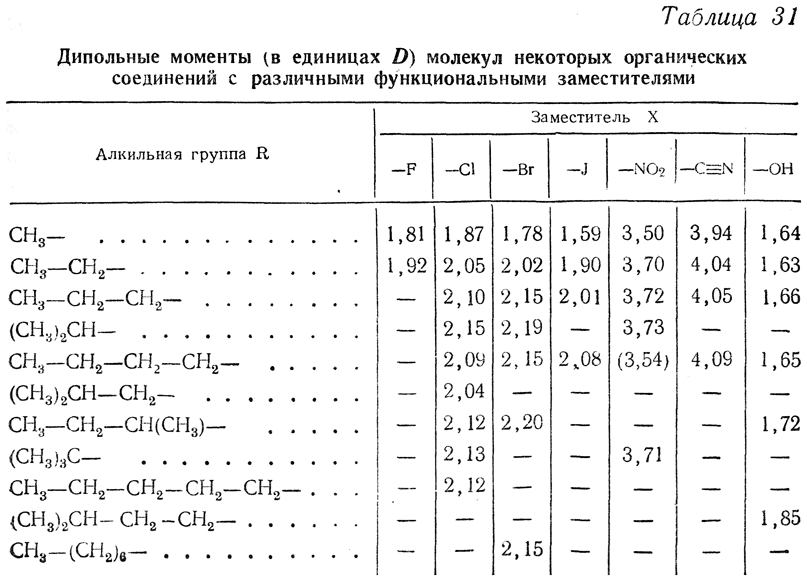
В более сложных молекулах надо, однако, учитывать и некоторые особенности. Так, например, поскольку для СН4 и ССl4 дипольный момент равен нулю, СН3Сl и СНСl3 должны были бы обладать одинаковыми дипольными моментами. Однако оказывается, что для хлористого метила СН3Сl эта величина (1,87 D) значительно больше, чем для хлороформа СНСl3, для которого μ=0,95D. Это может быть объяснено тем, что взаимное отталкивание трех ядер галоида в молекуле хлороформа сильно деформирует угол СlССl в сторону его увеличения (от 109° до ~116°), а следовательно, и углы НССl — в сторону их уменьшения.
Сопоставление дипольных моментов кислородных соединений
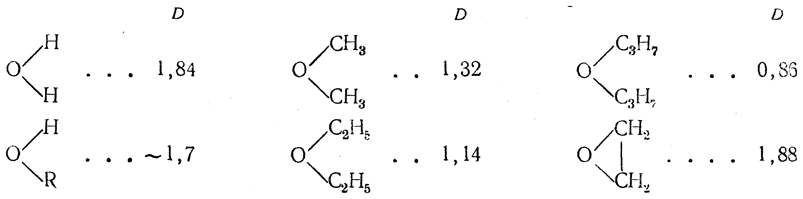
приводит к заключению, что угол между валентностями кислорода, составляющий у воды ~105°, все более и более деформируется в сторону увеличения в ряду простых эфиров, стремящихся, очевидно, приобрести энергетически наиболее выгодную конфигурацию, напоминающую конфигурацию парафинов (угол 112°).
В ряду спиртов R—О—Н такая симметризация, очевидно, не может быть достигнута ни при каком радикале R, чем и объясняется сравнительное постоянство дчпольного момента в этом ряду (μ≈l,7 D). В молекуле окиси этилена уменьшение валентного угла у кислорода (этот угол стремится стать близким к 60°) обусловливает увеличение дипольного момента, даже по сравнению с водой, до величины 1,88 D.
Линейные симметричные молекулы, вроде О=С=О, имеют μ=0 благодаря взаимной компенсации противоположно направленных сильных диполей связи С—О (μСО=2,5 D). Аналогичная компенсация диполей происходит, например, в случае дихлорзамещенных производных этилена. В го время как у 1,1-дихлорэтилена μ=1,7 D и у цис-1,2-дихлорэтилена μ=2,5 D, у транс-1,2-дихлорэтилена, имеющего центр симметрии, μ=0.
Для ряда кетонов дипольный момент составляет величину 2,5—2,7 D (для СН3—СО—СН3 μ=2,71D). Для ацетальдегида μ=2,72 D. Тример ацетальдегида, циклический паральдегид, в соответствии с большой симметрией, обладает дилолъным моментом, равным только 1,92 D.
Интересно отметить изменение дипольных моментов в ряду — аммиак, первичные, вторичные и третичные амины:

О том, как сказывается строение молекулы на величине дипольного момента при наличии кратных связей, дает понятие следующее сопоставление:
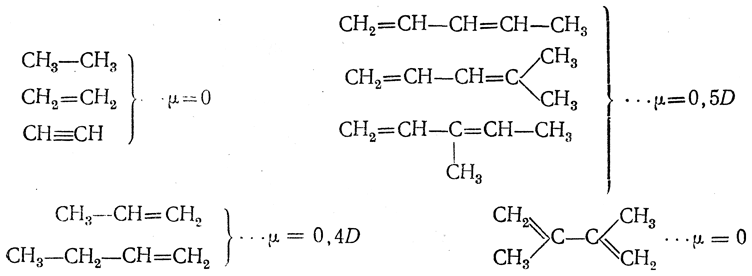
Интересные результаты дает определение дипольных моментов у 1,2-замещенных этанов:
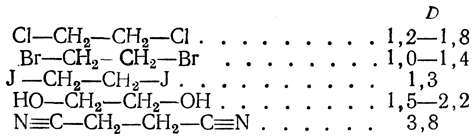
Для молекул этого типа возможно свободное, хотя и несколько ограниченное вращение вокруг оси С—С; очевидно, что при таком вращении дипольный момент, например, у Сl—СН2—СН2—Сl должен изменяться от некоторого максимального значения (при расположении атомов хлора друг над другом как бы в цис-положении, т. е. в заслоненной конформации) до нуля (при транс-положении атомов хлора, т. е. в заторможенной конформации).
Средний результирующий момент для дихлорэтана по расчету должен бы быть равен 2,4 D. На самом деле он много меньше, что подтверждает вытекающий из конформационных представлений вывод о преобладании заторможенной конформации. При повышении температуры, благодаря увеличению энергии вращения молекулы, вероятность возникновения энергетически менее выгодных конформации становится больше. Поэтому с повышением температуры у таких замещенных этанов дипольный момент увеличивается.
Дипольные моменты таких молекул, как С(СН2ОН)4, или С(ОС2Н5)4, или С(СН2СООСН3)4, которые, благодаря тождеству групп при тетраэдрическом атоме углерода, должны были бы быть разными нулю, оказывзются соответственно равными 2,0, 1,1 и 2,8 D. Это объясняется возможностью смещения векторов функциональных групп относительно направлений к вершинам тетраэдра.