Спектроскопия ЯМР
В отличие от масс-спектрометрии, мы не будем излагать здесь сколько-нибудь подробно физические основы спектроскопии ЯМР. Дело в том, что теория этого метода
и принципы работы приборов значительно сложнее, чем в масс-спектрометрии, а книга наша посвящена прежде всего сахарам. С другой стороны, по спектроскопии ЯМР имеется очень много доступных книг самого различного уровня – от популярных до весьма фундаментальных. Поэтому мы здесь ограничимся лишь самым поверхностным описанием спектроскопии ядерного магнитного резонанса, причем только резонанса на протонах (ПМР).
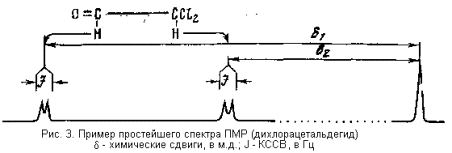
В обычном спектре ПМР каждый имеющийся в молекуле протон дает свой сигнал (другое дело, что сигналы двух или более протонов могут накладываться друг на друга). Этот сигнал характеризуется тремя параметрами: интенсивностью, которая прямо пропорциональна содержанию протонов данного типа в образце, величиной химического сдвига, измеряемого от некоторого внутреннего стандарта (чаще всего тетраметилсилана) в единицах м.д. (т.е. в миллионных долях рабочего поля прибора), и величиной константы спин-спинового взаимодействия с другим (или другими) протонами в той же молекуле. Эта константа, проявляющаяся в спектре в виде расщепления сигналов на отдельные компоненты и сокращенно называется обычно КССВ, измеряется в герцах (рис. 3).
Величина химического сдвига определяется многими особенностями структуры органической молекулы, а также рядом внешних факторов (в первую очередь применяемого для съемки спектра растворителя). Наибольший вклад в эту величину вносит плотность электронного облака вокруг данного протона: чем в большей степени электронная пара, образующая связь данного протона с ближайшим атомом, оттянута к этому последнему, тем большей будет величина химического сдвига δ . В очень грубом приближении можно сказать, что чем более кислым является данный протон, тем больше его химический сдвиг. Поскольку смещение электронов связи определяется в первую очередь природой включенного в эту связь атома, а во вторую – ближайшим окружением этого атома, величина химического сдвига отражает положение данного протона в молекуле и потому находит весьма многообразное применение в установлении структуры органических соединений.
Константа спин-спинового взаимодействия описывает спиновую связь данного протона с другими протонами в той же молекуле. Это взаимодействие осуществляется только по цепи ковалентных связей и поэтому быстро ослабевает с увеличением расстояния между взаимодействующими ядрами. Обычно приходится сталкиваться с КССВ протонов через две связи (в группах H-C-H) и через три связи (в группировках H-C-C-H). Величины КССВ также определяются многими структурными факторами. Главным из них является геометрия соответствующего фрагмента. Именно на этом обстоятельстве в первую очередь основано применение спектроскопии ПМР для установления конфигурации ассиметрических центров в сахарах. Геометрия фрагмента H-C-H, даже будучи надежно установлена, дает мало интерсных сведений о структуре и конфигурации изучаемого соединения. Поэтому соответствующая ей КССВ играет подчинительную роль в применении ПМР-спектров сахаров (кроме того, таких фрагментов в углеводах всегда мало). Напротив, геометрия фрагментов H-C-C-H в совокупности определяет всю стереохимию типичной моносахаридной молекулы. Поэтому измерение соответствующих КССВ и их интерпретация играют первостепенную роль в структурном применении спектроскопии ПМР.
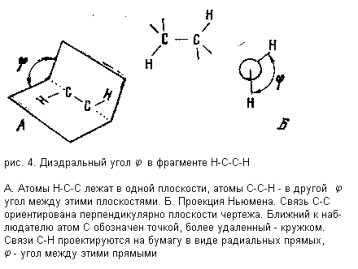
Если (в первом приближении) считать длины связей и углы H-C-C и C-C-H постоянными величинами, то геометрия фрагмента H-C-C-H может быть описана одним параметром: так называемым диэдральным, или двугранным, углом j , смысл которого виден из рис. 4.
Величина КССВ двух протонов в фрагменте H-C-C-H зависит главным образом от диэдрального угла. К сожалению, однако, КССВ зависит не только от этого
угла, но и от ряда других структурных особенностей молекулы. Поэтому все математические описания зависимости КССВ только от угла j носят неизбежно приближенный характер (таково, например, широко известное уравнение Карплюса). Тем не менее почти всегда справедливо следующее грубое правило: Если диэдральный угол приближается к 0 ° или 180 ° , то КССВ – «большая», т.е. составляет около 8-10Гц; если этот угол близок к 90 ° , то КССВ близка к 0, если же (и это весьма распространенный случай в реальных структурах) этот угол близок к 60 ° , то КССВ бывает «малая», т.е. порядка 1-3 Гц. Несмотря на явно приближенный характер этого правила, оно находит очень широкое и плодотворное применение в структурных исследованиях. Вот типичный пример из химии углеводов.
Требуется определить конфигурацию гликозидного центра в тетраацетате D-глюкопиранозида, т.е. сделать выбор между двумя структурами: 34а и 34б. Анализ основан на КССВ протона при С-1 с протоном при С-2 (этот фрагмент выделен на схеме жирными линиями). В первом случае ( α -глюкозид) диэдральный угол H-C-C-H составляет около 60 ° , т.е. для него следует ожидать малой КССВ. Для β -аномера (34б) этот угол близок к 180 ° (Это два аксиальных заместителя, их связи параллельны оси цикла). Поэтому для такого соединения КССВ протона при С-1 должна быть большой. Наблюдаемая величина КССВ (8 Гц) позволяет уверенно сделать выбор в пользу структуры 34б.
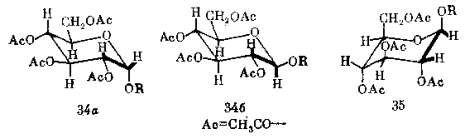
Внимательный анализ рассмотренного примера показывает, что здесь спектроскопия ПМР позволяет одновременно решить вопрос и о конформации изучаемого соединения, и о конфигурации ассиметрического центра. Точнее говоря, вопрос о конфигурации спектроскопией ПМР в данном случае не решается: она определяется лишь опосредованно, через конформацию. В самом деле, глюкозиду 34б можно, по крайней мере, формально, приписать конформацию 35. В этом случае диэдральный угол фрагмента H-C-C-H составлял бы уже не 180 ° , а около 60 ° , ожидаемая КССВ была бы существенно иной, а выводы из спектра ПМР могли бы оказаться прямо ошибочными. Однако из общих принципов конформационного анализа мы можем заключить, что для β -глюкозида конформация 35, если и реализуется, то в достаточно малой пропорции по сравнению с конформацией 34б. Так что сделанное выше заключение остается в силе.
Таким образом, можно видеть, что для эффективного применения спектроскопии ПМР в решении структурных задач в сахарах, т.е. для установления конфигураций, необходимо по возможности уменьшить неопределенность, связанную с возможными конформационными состояниями изучаемых молекул. Прежде всего для этого применяют циклические производные, например гликозиды, и избегают применять ациклические, типа полиолов, в которых конформационные возможности значительно шире. Обычно стремятся применять такие производные и в таких растворителях, для которых конформационные закономерности наиболее просты и однозначны. В этом смысле, например, пиранозные формы сахаров явно предпочтительнее фуранозных, а ацетаты сахаров предпочтительны перед свободными сахарами. К сожалению, дать этому трактовку, не вдаваясь в подробности конформационного анализа, не представляется нам возможным.
В примере с глюкозидами мы показали, что знание КССВ протона при C-1 позволяет сделать надежное заключение о конфигурации ассиметрического центра С-1. И это вполне справедливо при тех условиях задачи, которые мы сформулировали выше: установить аномерную конфигурацию D-глюкопиранозида, т.е. гликозида, для которого известна конфигурация всех остальных центров асимметрии. Однако, если бы мы не знали конфигурации остальных центров, то полученный результат говорил бы лишь об относительной конфигурации двух соседних центров: С-1 и С-2, а не об их абсолютных конфигурациях, что означало бы существование двух вполне равноправных вариантов ответа. Дальнейший анализ спектра показал бы нам, что протон при С-2 имеет, в свою очередь, большую КССВ по сравнению со следующим соседом, последний – протон при С-3 – имеет также большую КССВ с протоном при С-4, а протон при С-4 – большую КССВ с протоном при С-5. Такой набор данных позволил бы заключить, что в изучаемом соединении все протоны пиранозного цикла аксиальны, так как только при этом условии все КССВ этих протонов могли оказаться большими. Это прямо означает, что все заместители в пиранозном цикле экваториальны, т.е. автоматически приводят к глюко-конфигурации (правда, опять относительной: спектроскопия ПМР принципиально не способна отличать оптические антиподы). Таким образом, подробный анализ спектра глюкозида 34б привел бы к четким выводам об относительной конфигурации всех ассиметрических центров, т.е. к решению фишеровской задачи. Именно на таком анализе спектров и основано в первую очередь определение конфигурации моносахаридов с помощью спектроскопии ПМР.
Спектр глюкозида 34б очень прост в том смысле, что все КССВ большие, следовательно все протоны аксиальны. Более того, этот случай уникален. При любой другой конфигурации, какие-то КССВ окажутся малыми, а какие-то большими. Когда нам известно, какая КССВ к какой паре протонов относится, задача решается более или менее просто. Однако на пиках в спектре отнюдь не написано, какому протонув молекуле принадлежит данный сигнал. Мало того, совсем не исключение, а скорее правило, что в спектрах производных углеводов сигналы части протонов накладываются друг на друга. Поэтому главной трудностью, с которой сталкивается исследователь при анализе спектра ПМР сахаров, является отнесение сигналов, т.е. установление взаимооднозначного соответствия определенного сигнала в спектре и определенного протона в молекуле. Как же решается такая задача?
Надо сразу сказать, что стандартных путей к решению здесь нет. Точнее говоря, путь есть и совершенно надежный и общий; однако его применение сопряжено с весьма значительными затратами труда и времени. Он состоит в синтезе дейтерированных аналогов изучаемого соединения и сопоставлении спектров аналогов и родоначального вещества. Идея этого подхода основана на том, что дейтерий в спектрах ПМР не виден (его химический сдвиг лежит далеко за пределами шкалы химических сдвигов протонов). В результате в спектре дейтерированного аналога исчезает сигнал одного из протонов, а именно того, который был заменен на дейтерий. И по этому признаку можно с полной надежностью произвести отнесение такого сигнала. Имея серию дейтерированных производных, можно полностью расшифровать весь, сколь угодно сложный, спектр ПМР.
По-видимому, даже непосвященному в таинства органического синтеза ясно, что такой путь очень трудоемок и не может применяться в качестве рутинного метода хотя бы по той простой причине, что для осмысленного планирования синтеза дейтерированных аналогов нужно прежде всего знать структуру соединения, а это лишает смысловой основы расшифровку спектра ПМР как шага на пути установления структуры соединения. Поэтому синтез дейтероаналогов применяется тогда, когда расшифровка сложных спектров имеет самодовлеющее значение, например в исследовании закономерностей спектра ПМР новых классов соединений и т.д. (собственно, закономерности, на которые мы теперь опираемся при структурном примененииПМР, и были в свое время добыты таким трудоемким путем). В рутинном же применении ПМР для структурных исследований отнесение сигналов в значительной мере основывается на изученных ранее особенностях спектров соединений этого класса, на ряде общих закономерностей спектроскопии ПМР, а также на многих частных приемах расшифровки.
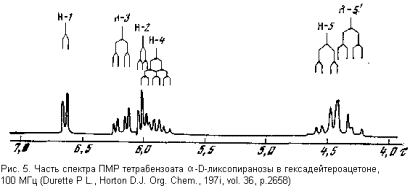
Для иллюстрации приведем здесь спектр тетрабензоата α -D-ликсопиранозы (рис. 5) и его трактовку. При этом мы упростим свою задачу: не будем расшифровывать шаг за шагом спектр, полученный с прибора, а попытаемся разобраться, и то лишь в общих чертах, почему сигналы определенных протонов расположились в спектре именно таким образом (будем рассматривать только наиболее информативную часть спектра – протоны пиранозного цикла).
Мы уже упоминали, что главный вклад в величину химического сдвига вносит электронная плотность вокруг данного ядра. В рассматриваемом соединении наименьшая электронная плотность должная окружать протон при С-1. В самом деле, у этого углеродного атома имеется два кислородных, оттягивающих электроны заместителя. Поэтому сигнал протона Н-1 вполне закономерно оказывается в спектре в самом слабом поле, т.е. имеет наибольший химический сдвиг. Наоборот, каждый из протонов при С-5 испытывает влияние другого протона, связанного с тем же углеродным атомом. Связь C-H поляризована таким образом, что ее электроны несколько смещены к углероду. Поэтому электронная плотность вокруг протонов при С-5 несколько выше, чем вокруг любого другого протона в молекуле. В соответствии с этим оба протона имеют наименьший химический сдвиг (располагаются в наиболее сильном поле). Электронное окружение протонов при С-2, С-3 и С-4 весьма сходно, и неудивительно, что их сигналы располагаются в спектре близко друг к другу. В отнесении этих сигналов могут помочь следующие соображения.
Спин-спиновое взаимодействие двух протонов вполне взаимно: оно приводит к расщеплению сигналов каждого из взаимодействующих протонов в дублет, и, конечно, с одной и той же КССВ. Если с данным протоном связан спиновой связью еще один, то взаимодействие с ним осуществляется независимо от взаимодействия с первым партнером. Таким образом, каждый компонент дублета, возникшего в результате взаимодействия с первым протоном, расщепляется на дублет со свой КССВ. В результате протон, взаимодействующий с двумя другими, дает сигнал в виде квартета (или, точнее, дублета дублетов). Знание этих особенностей (мы снова подчеркиваем, что ни слова не говорим о физике явления, а рассматриваем тот крайний минимум сведений, который можно использовать, так сказать, потребительски) играет большую роль в расшифровке спектров ПМР.Теперь вернемся к нашему примеру.
Протон при С-1 уникален в том отношении, что на расстоянии не более трех ковалентных связей от него находится только один другой протон – при С-2 (у остальных протонов кольца имеется не менее двух таких соседей). Поэтому его сигнал должен быть дублетом, как оно и есть на самом деле. По этому признаку мы могли бы отличить его от всех остальных, даже не прибегая к соображениям химического сдвига. Его взаимодействие с протоном Н-2 описывается КССВ 3,1 Гц. Следовательно, такая же КССВ должна быть у сигнала протона Н-2. Просмотрев остальной спектр, мы обнаруживаем сигнал, у которого одна из КССВ составляет 3,1 Гц и отнесем его к протону при С-2. Измерив его вторую КССВ(3,3 Гц), мы узнем о взаимодействии с протоном при С-3. Аналогично находим и сигнал протона при С-3 (по признаку КССВ 3,3 Гц), и у него обнаруживаем вторую (большую) КССВ (9,0 Гц) с протоном при С-4 и т.д.
Наша задача не в том, чтобы научить читателя методике расшифровке спектров ПМР (в этом смысле изложенное выше весьма схематично), а в том, чтобы по возможности передать логику мышления в этой области. И в связи с этим особенно важно обратить внимание на два обстоятельства. Первое. В наших рассуждениях мы опирались на знаение структуры изучаемого соединения – мы могли не знать его стереохимии, но на бутлеровскую структуру ссылались постоянно. В этом смысле спектроскопия ПМР дает (в некоторых пределах, конечно) тем больше новых сведений об изучаемом соединении, чем большей информацией о нем исследователь уже располагает. Для изучения структуры «с нуля» метод ПМР часто мало эффективен. Второе. Отнесение сигналов по цепи спиновых связе опирается на отнесение одного (первого) сигнала. Ошибка в первом отнесении неизбежно приведет к полностью ошибочной интерпретации всего спектра. Некоторой гарантией верности отнесений может служить логическая увязка отнесений всех сигналов в спектре как с точки зрения спиновых связей, так и с точки зрения имеющихся сведений о структуре. Поэтому очень рискованно бывает делать серьезные выводы из спектра ПМР, в котором удалось отнести лишь часть сигналов.
Не вдаваясь в подробности, укажем в заключение, что мощным подспорьем в расшифровке спектров ПМР служит такие инструментальные методы, как двойной резонанс и ИНДОР, позволяющие объективно установить наличие спиновых связей между ядрами; динамический ЯМР (ДЯМР), позмоляющий устранить неопределенности, связанные с конформационными равновесиями, а также метод сдвигающих реагентов (или, как их часто называют на английский манер, шифт-реагентов), с помощью которого можно избирательно и весьма сильно изменить химические сдвиги отдельных протонов (как говорят на лабораторном жаргоне, «вытянуть их из каши», т.е. из группы перекрывающихся и потому почти не поддающихся интерпретации сигналов).
Нам бы не хотелось, чтобы у читателя сложились абсолютистские представления о современных методах установления строения моносахаридов. Ну, например, такие: есть два стандартных метода – масс-спектрометрия и спектроскопия ПМР, которые позволяют автоматически устанавливать строение любых моносахаридов (стоит только им научиться и получить доступ к приборам). Или иначе: без масс-спектров и спектров ПМР структуру не установить. И то и другое неверно.
Конечно, оба эти метода – исключительно мощные инструменты исследования. Однако это отнюдь не «черные ящики», где на входе вещество,а на выходе готовая структура. На выходе – всего лишь спектр, а структура появляется в результате интерпретации спектра. Последняя же отнюдь не трафаретна и требует от исследователя (именно от самого исследователя, а не от того, кто управляет прибором и выдает спектры) больших знаний, опыта, интуиции. Помимо спектроскопии, современная химия углеводов располагает целым комплексом точных и тонких методов структурного анализа, которые, хотя и не опираются на новейшие приборы, позволяют делать не менее надежные заключения о структуре. Бывает так, что самыми примитивными, известными с прошлого века «пробирочными» пробами можно узнать о структуре моносахарида не меньше, чем используя самую совершенную аппаратуру. Мы, конечно, далеки от того, чтобы пропогандировать идею возврата кэпохе жаровен и реторт, но хотим подчеркнуть широту и многообразие накопленного к настоящему времени арсенала методов структурных исследований. И в оценке той или иной работы самую последнюю роль должны играть соображения новизны примененных методов или, тем более, их модности.
Несравненно важднее, чтобы метод (пришедший ли из прошлого века или из самоновейшей литературы) был применен к месту, чтобы он позволял делать максимально надежные заключения с минимальными затратами труда. И никакой самоновейший метод при бездумном применении не гарантирует от грубых ошибок.
Короче говоря, установление строения моносахаридов как почти любое другое структурное исследование, есть творчество. В нем могут применяться весьма совершенные и разработанные методы исследования, приемы простые и сложные, но их совокупность, последовательность и ход мысли исследователя ни в какие стандартные правила не укладываются.
Это и есть творческая деятельность, которая, как известно, отличается от прочих тем, что для нее нельзя составить алгоритма или программы. Так что, в конечном счете, методы суть лишь инструменты, а струтуры устанавливаются не методами, а интеллектуальными усилиями ученых.