Масс-спектрометрия
Итак, имеется моносахарид или его метилированное производное. Установить строение – значит решить две группы задач. Прежде всего надо выяснить длину углеродной цепи, природу, число и расположение функциональных групп; для метилированных сахаров, в частности,- число и положение метильных групп. Все это в совокупности иногда называют бутлеровской структурой. Затем нужно установить конфигурацию ассиметрических центров, т.е. решить задачу того же типа, которую решал Эмиль Фишер для глюкозы, маннозы и арабинозы. В этой главе мы рассмотрим пути решения задач первой группы одним наиболее общим и употребительным в современной науке методом – с помощью осколочной масс-спектрометрии.
Принципиально масс-спектрометр состоит из четырех блоков: системы напуска, ионного источника, системы магнитной фокусировки и детектора (рис. 1). В системе напуска образец анализируемого вещества испаряют в вакууме. Образовавшиеся пары поступают в ионный источник, где подвергаются бомбардировке пучком ускоренных электронов (энергия обычно порядка десятков электронвольт). Энергия облучения расходуется на выбивание электронов из молекул анализируемого вещества – последние превращаются в положительно заряженные ион-радикалы. Такие частицы высоко реакционноспособны и нестойки. Тут же в ионизационной камеры они претерпевают распад на заряженные и незаряженные осколки (отсюда название метода «осколочная масс-спектрометрия»). Вся ионизационная камера находится под высоким положительным
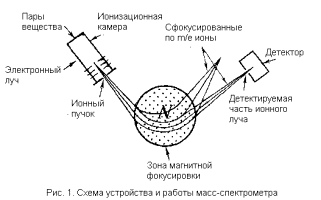
потенциалом по отношению к остальным частям прибора. Поэтому электростатическое поле выталкивает из камеры положительные ионы. Перед выходом из камеры пучок ионов проходит через систему электростатических линз и диафрагм, так что в результате из камеры выходит узкий сфокусированный ионный луч, в котором скорости ионов зависят от их масс и зарядов.
Ионный пучок далее попадает в зону магнитной фокусировки. Здесь в магнитном поле прямолинейные траектории ионов искривляются, причем геометрия магнитного поля рассчитана так, чтобы сфокусировать ионы на детекторе. В конечном итоге ионы подходят к детектору по индивидуальным траекториям, которые целиком определяются величиной отношения массы иона к его заряду (m/e). Варьируя электростатическое или магнитное поле, можно сфокусировать на детекторе ионные потоки для каждого значения m/e и измерить количественно соответствующий таким частицам ионный ток, т.е. величину, пропорциональную числу частиц с данным m/e в анализируемой плазме. Развертка по m/e дает масс-спектр, в котором по оси абсцисс отложены величины m/e, а по оси ординат – интенсивности ионного тока, или, что то же самое, доля частиц с данным m/e в плазме (рис. 2). Поскольку в подавляющем большинстве случаев образующиеся осколки однозарядны, шкала m/e практически совпадает со шкалой ионных масс.
В описанных условиях масс-спектрометрия (а они самые обычные, но далеко не единственные) органические
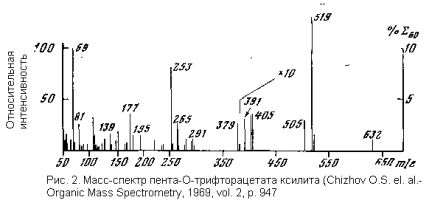
вещества дают сложные масс-спектры. В них, однако, удается выделить наиболее характерные и наиболее интенсивные пики, отвечающие главным путям распада изучаемого соединения. Поскольку типичные пути распада многих классов органических соединений, в частности моносахаридов, сейчас подробно изучены, по картине масс-спектра можно составить достаточно полное представление о структуре изучаемого соединения, затратив на это минимум вещества (меньше миллиграмма, нередко лишь микрограммы) и минимум времени (на съемку спектра на хороших приборах требуются считанные минуты; иное дело, что расшифровка спектра может занять несравненно больше времени).
Как же расшифровывают масс-спектры? «Читают» спектр обычно справа налево – от больших масс к малым. И это не прихоть: крупные осколки обычно наиболее информативны. Для них возможно лишь весьма ограниченное число путей образования, тогда как «мелочь» может возникать самыми разными путями и из нее извлечь аналитически полезную информацию гораздо труднее.
Первый пик в спектре – пик молекулярного иона, т.е. ионизированной, но не распавшейся исходной молекулы. При описании спектра его обозначают буквой М. Уже из этого пика можно извлечь много полезных сведений. В самом деле, молекулярные массы – это не температуры плавления или удельные вращения. Они могут иметь только дискретные значения, подчиняющиеся вполне определенным закономерностям. Ну, например, таким простейшим, как то, что любое соединение состава C n H m O p может иметь только четную молекулярную массу. Значение молекулярной массы сразу резко ограничивает число возможных структур, а более подробный анализ спектра в области пика молекулярного иона позволяет получить еще целый ряд дополнительных данных. Мы здесь не будем разбирать этот аспект, а укажем лишь на один характериный пример. Природный бром состоит из двух изотопов 79 Br и 81 Br в соотношении 1:1. Поэтому молекулярный ион любого соединения, содержащего один атом брома, дает в масс спектре два пика равной интенсивности, различающиеся на две единицы массы. Такой дуплет в спектре весьма характерен и сразу указывает на наличие в анализируемом соединении одного атома брома. А если бы в нем было два атома брома, то соответствующие ионы дали бы пик в виде триплета с расстоянием между компонентами в две единицы массы и соотношением интенсивностей 1:2:1.
Дальше по спектру идут пики осколков. Молекулярный ион распадается на две частицы: заряженную и нейтральную. Последняя часто оказывается высокоустойчивой малой молекулой типа H 2 O, CO и т.п. Эти фрагменты нейтральны, однако их можно идентифицировать косвенно – по разности масс молекулярного иона и заряженного осколка. Поэтому последние часто описывают в разностном выражении, например:M-H 2 O, или M-18; M-CO, или M-28; M-CH 3, или M-15; M-H 2 C=C=O, или М-42 и т.д. Состав таких больших осколков обычно легко идентифицировать, так как число вариантов состава малых осколков весьма невелико. Так, например, для обычных органических соединений М-18 – это всегда M-H 2O. Таких первичных осколков, т.е. тех, которые возникают непосредственно за распадом молекулярного иона, может быть несколько, так как распад может протекать по нескольким направлениям. Первичные осколки, в свою очередь, подвергаются распаду, а продукты распада тоже распадаются. Так возникают серии ионов, отвечающих определенным путям распада, или, как чаще говорят, фрагментации молекулярного иона. Искусство расшифровки спектра в значительной мере состоит в умении из большого числа пиков выделить такие, которые увязываются в определенные серии – последовательности фрагментации исходного иона. Когда такие серии выявлены, восстановить картину распада и, следовательно, структуру анализируемого вещества уже значительно проще, особенно если исследователь опирается на общие данные о характерных путях фрагментации соединений данного класса.
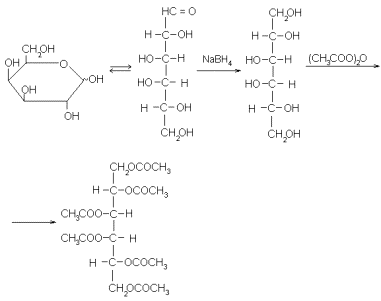
Теперь, наконец, можно уже конкретно перейти к масс-спектроскопии моносахаридов. Непосредственно исследовать их этим методов затруднительно. Дело в том, что молекулы моносахаридов содержат много полярных групп, а это самым неблагоприятным образом сказывается на их летучести. Выход из положения состоит в получении подходящих более летучих производных. На их выбор накладывается целый ряд ограничений, но к настоящему времени эта трудность уже преодолена: найдено несколько классов производных, отвечающих всем требованиям, и подробно изучены закономерности их фрагментации. Чаще всего для этой цели сейчас используются ацетаты полиолов. Их получают с помощью двух весьма общих и чрезвычайно простых в экспериментальном оформлении реакций: восстановление моносахарида боргидридом натрия и последуюшего ацетилирования. Ниже эти реакции показаны на примере D-галактозы (с. 71).
Фрагментация ацетатов полиолов такого типа относительно проста. Она включает первичные ионы, образующиеся путем разрыва C-C связей углеродного скелета, а также отщепления CH 3 COO. Зная массу таких осколков и массу молекулярного иона, легко составить представление
о структуре исходного полиола и, следовательно, моносахарида. Если в молекуле имеется дезоксизвено, как например в ацетате 28, образующемся из 2-дезокси-D-рибозы (27), то характерным оказывается разрыв цепи в β -положении к дезоксизвену. Зная эту закономерность, по величине m/e соответствующих фрагментов (в данном случае m/e 159) можно установить положение дезоксизвена (см. с. 72).
Тут, однако, возникает одна неопределенность. Действительно, ацетат 29, образующийся из изомерной 4-дезоксирибозы 30, будет давать точно такой же фрагмент с m/e 159, только из нижней части молекулы. Поэтому мы еще не вправе приписать структуру исходному сахару – это может быть либо 2-дезоксипентоза, либо 4-дезоксипентоза.
Выход, однако, есть. Надо искусственно внести асимметрию в молекулу. Для этого вместо боргидрида натрия на стадии восстановления применяют его изотопный аналог – тетрадейтерийборгидрид. Тогда у углеродного атома бывшей карбонильной группы, т.е. при С-1, появляется один атом дейтерия вместо одного атома протия. В результате ацетат полиола 31 из 2-дезокси-D-рибозы дает в масс спектре тот же характеристический пик, но сдвинутый на единицу массы (разница масс дейтерия и протия), т.е. пик иона с m/e 160 вместо 159. А ион, возникающий
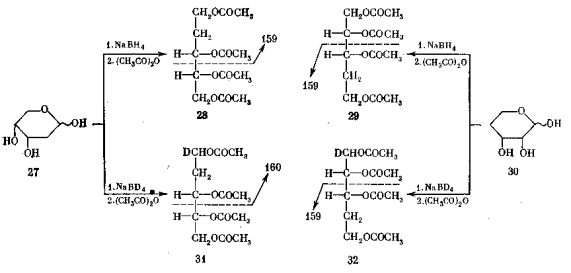
путем аналогичной фрагментации из изомерного соединения 32, дейтерия не содержит и, следовательно, будет по прежнему иметь m/e 159.
Для метилированных моносахаридов аналогичное превращение дает частично метилированное и частично ацетилированные полиолы. Характерное направление фрагментации таких производных – это разрыв C-C-связи около метоксильной группы. Особенно характерен такой разрыв между двумя соседними метоксильными группами (если такая пара в молекуле имеется):
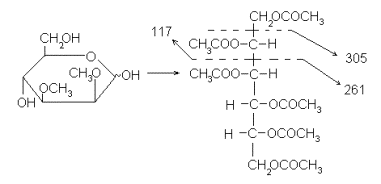
По таким фрагментам можно установить положение метоксильных групп, т.е. решить главную задачу, возникающую при структурном исследовании метилированных моносахаридов. Неопределенность, возникающая здесь из-за относительно высокой их симметрии (т.е. либо 2,3-ди-О-метил-, либо 4,5-ди-О-метил-) может быть легко устранена аналогично тому, как описано выше, т.е. с применением дейтероборгидрида на стадии восстановления. Вообще надо сказать, что введение изотопной, особенно дейтериевой, метки – весьма распространенный прием в масс-спектрометрии. Вот, например, как была доказана бутлеровская структура 4-О-метил-D-глюкуроновой кислоты, полученной синтетически в виде метилового эфира 33:
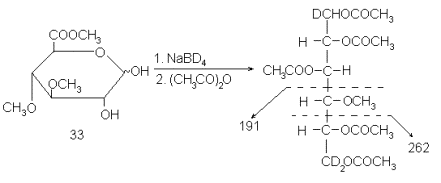
Здесь из масс-спектра, точнее, всего из двух пиков ионов с m/e 262 и 191, следовала сразу вся структура – число ацетильных и метильных групп, а также тот факт, что в молекулу при восстановлении вошло три атома дейтерия, т.е. что исходное соединение было эфиром гексуроновой кислоты (один дейтерий входит при восстановлении альдегидной группы, а два – при восстановлении этерифицированного карбоксила). Кроме того, пики ионов с m/e 191 и 262 однозначно определяют положение метильной группыи и без всяких неопределенностей, так как концы были помечены дейтерием, причем по-разному: у С-1 – один атом, а у C-6 – два.
Итак, масс-спектрометрия – чрезвычайно информативный метод установления строения. Но для нее, конечно, нужно иметь индивидуальное вещество, т.е. произвести предварительное разделение смеси, в которой вещество находится.Такой результат достигается непросто (особенно при работе с метилированными сахарами) требует сложной (и в экспериментальном и в приборном отношении) хроматографической техники. Наивысшее современное достижение в этой области – объединение газо-жидкостного хроматографа и масс-спектрометра в одном приборе, т.е. анализ смеси методом, получившем название хромато-масс-спектрометрии.
В газо-жидкостном хроматографе вещество вносят в колонку – длинную узкую трубку с нелетучей жидкой фазой, нанесенной на пористый инертный твердый материал. Через колонку пропускают струю газа-носителя при определенной регулируемой температуре. Вещество в виде паров движется по колонке с током газа, непрерывно подвергаясь распределению между газовой (подвижной) и жидкой (неподвижной) фазами. Время, в течение которого данное вещество проходит колонку (так называемое время удерживания) зависит от летучести вещества и его способности абсорбироваться данной жидкой фазой. Оба свойства определяются тонкими особенностями структуры конкретного соединения, так что время удерживания весьма характерно и индивидуально для каждого вещества в конкретных условиях разделения. Поэтому, если в колонку внесена смесь веществ, то ее компоненты появляются на выходе из колонки в разное время: достигается их разделения. После выхода их колонки газовый поток попадает в детектор, регистрирующий появление вещества, а сигналы с детектора через усилительную схему поступают на самописец. В результате самописец выписывает кривые выхода вещества из колонки в зависимости от времени, т.е. рисует газо-жидкостную хроматограмму. Таким образом можно проанализировать состав весьма сложных смесей веществ.
Идея хромато-масс-спектрометрии состоит в том, что газы и пары, прошедшие колонку, вводят в ионизационную камеру масс-спектрометра, объединенного с хроматографом в единый комплекс – хромато-масс-спектрометр. В результате исследователь получает из одного анализа смеси сведения о временах удерживания ее компонентов, об их относительном содержании в смеси и, наконец, тут же получает масс-спектры каждого компонента смеси.
Такой метод анализа идеально подходит для изучения смесей метилированных сахаров, получающихся при мономерном анализе полисахаридов с помощью метода метилирования. В самом деле, хромато-масс-спектрометрия позволяет идентифицировать известные вещества со свидетелями при помощи прямого сравнения и тут же, используя масс-спектрометр, дополнительно подтверждать их структуру, а для неизвестных веществ или для тех, для которых не оказалось нужного заведомого образца,- установить строение (без конфигураций, конечно) по масс-спектру.
В настоящее время хромато-масс-спектрометрия – магистральный путь развития структурного анализа полисахаридов, позволяющий получить на нескольких миллиграммах изучаемого биополимера за считанные дни информацию, для добывания которой еще совсем недавно требовались десятки, а то и сотни граммов материала и годы труда.