Синтез моносахаридов
Защитные группы
Сама по себе идея применения защитных групп широко известна в общей органической химии. Вот классический пример. Нужно пронитровать анилин и получить п-нитроанилин. Азотная кислота – сильный окислитель, а аналин легко окисляется. Следовательно, нитровать его непосредственно нельзя. Поэтому аминогруппу анилина предварительно защищают: превращают в ацетат, гораздо более устойчивый к окислителям, затем нитруют и в заключение удаляют защиту с аминогруппы щелочным гидролизом:
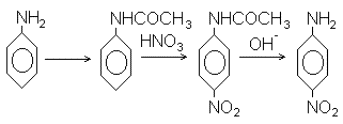
Здесь все просто. Анилин содержит два весьма различных по характеру реакционных центра – аминогруппу и ароматическое ядро. Поэтому избирательно защитить один из них не составляет проблемы. Продукт реакции – п-нитроанилин – весьма устойчивое соединение и легко переживает условия достаточно жесткого щелочного гидролиза. Следовательно, удаление защиты также не вызывает затруднений. В химии углеводов дело обстоит несравненно сложнее. Прежде всего, здесь функциональные группы весьма сходны, так что ввести защиту избирательно – а в этом весь смысл такой операции – весьма непросто. Таких групп в молекуле несколько (чтобы не сказать много), а защитить нужно все, кроме одной-двух. Понятно, что это обстоятельство, вообще говоря, не упрощает задачу. Наконец, сами углеводы и практически все их производные – соединения достаточно высоко реакционноспособные. Из-за этого возможности воздействий, пригодных для удаления защит на заключительных стадиях, а следовательно, типы применяемых защитных групп жестко ограничены.
Основные требования к защитным группым достаточно очевидны. Во-первых, они должны допускать избирательное введение. Во-вторых, сами защиты должны быть вполне устойчивы в условиях основной реакции. В-третьих, защиты должны допускать удаление в условиях, обеспечивающих сохранность как самой углеводной структуры, так и, разумеется, результатов главной реакции, ради осуществления которой и возводились защитные сооружения. Наконец, не столь принципиально, но весьма немаловажно, чтобы реакции введения и удаления защитных групп проходили с высокими выходами: иначе весь многостадийный синтез будет сопряжен со слишком значительными потерями.
Из всего перечисленного наибольшие затруднения вызывает избирательное введение. Здесь нет каких-то разработанных правил, следуя которым можно механически выбрать необходимую последовательность превращений и типы защитных групп. Тем не менее есть ряд хорошо разработанных реакций, ведущих к образованию защит, и ряд принципов обеспечения их региоспецифичности. Так что сейчас грамотый синтетик может составить реальный план синтеза, ведущего к избирательному освобождению любой функциональной группы в любом моносахариде. Но, подчеркнем еще раз, это не механическое применение готовых правил, а творческий процесс, требующий тщательного учета задач конкретного синтеза и выбора оптимальной схемы из ряда возможных. Поэтому не будем пытаться дать, так сказать, алгоритм для избирательной защиты функций, а опишем лишь некоторые элементарные приемы, применяемые в химии углеводов для этой цели.
Рассмотрим D-глюкозу (2). Пусть нам надо защитить все гидроксильные группы, кроме гидроксила при С-6. Такая задача сравнительно проста, так как интересующий нас гидроксил первичный и заметно отличается по реакционной способности от остальных гидроксилов в молекуле – вторичных спиртовых и полуацетального. Эту повышенную реакционную способность и используют на ключевой стадии синтеза. Глюкозу обрабатывают трифенилметилхлоридов (тритилхлоридом, как его часто сокращенно называют) в пиридине. При реакции тритилхлорида со спиртами образуются простые тритиловые эфиры. Тритильная группа весьма объемиста, поэтому тритилирование пространственно более затрудненных вторичных спиртов проходит медленно, тогда как первичные тритилируются легко. Благодаря этому тритилирование глюкозы проходит с высокой избирательностью и ведет к образованию тритилового эфира 12. Все остальные гидроксилы можно далее защитить ацетилированием уксусным ангидридом в пиридине. В полученном производном 13 все функциональные группы защищены, но защищены по-разному. Тритиловый эфир может быть разрушен кислотным гидролизом в таких условиях, которые не затрагивают сложные эфиры – ацетаты. Продуктом такого гидролиза является тетраацетат 14, в котором свободен единственный гидроксил – при С-6.
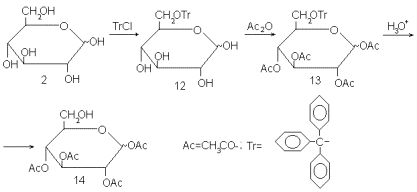
Обратите внимание, каким парадоксальным путем идет этот синтез: для того, чтобы избирательно освободить гидроксил при С-6, мы начинаем с того, что его защищаем. И тем не менее конечная цель достигается весьма успешно. Пример характерен в двух отношениях: во-первых, химия углеводов в части логики введения избирательных защит полна таких парадоксов, а во-вторых, использование избирательного тритилирования является общим (что редко в этой области) методом освобождения первичного гидроксила в сахарах.
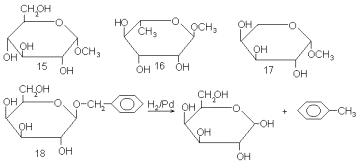
Другой участок в молекуле моносахарида, также обладающий специфическими свойствами,- это гликозидный центр. Для его избирательной защиты чаще всего применяют синтез низших гликозидов, в простейшем случае путем катализируемой кислотами конденсации моносахаридов со спиртами (синтез гликозидов по Фишеру). Наиболее распространенные производные для этой цели – метилгликозиды, каковы, например α -метил-D-глюкопиранозид (15), α -метил-L-рамнопиранозид (16) или β -метил-L-арабинопиранозид (17). Для расщепления метил-гликозидов необходимо произвести достаточно жесткий кислотный гидролиз или ацетолиз, что не всегда приемлемо по условиям устойчивости основного продукта. Чтобы избежать этого осложнения, пользуются бензил-гликозидами [например, β -бензил-D-галактопиранозидом (18)], в которых защита может быть удалена в специфических условиях путем гидрогенолиза над палладиевым катализатором (см. схему).
Наибольшие трудности возникают при необходимости избирательной защиты части вторичных гидроксилов моносахаридов, так как эти группы обладают наиболее близкими химическими свойствами. Чаще всего ключевой стадией в таких синтезах является образование тех или иных ацеталей или кеталей. Как известно, альдегиды и кетоны способны легко конденсироваться со спиртами в присутствии кислотных катализаторов с образованием ацеталей или кеталей 19. Если в реакцию вводят двухатомный спирт с подходящим расположением гидроксильных групп, то такая реакция приводит к аналогично построенным циклическим производным типа 20. Ацетали и кетали расщеаляются кислотным гидролизом в сравнительно мягких условиях и весьма устойчивы к щелочам, что делает их пригодными в качестве защитных групп в многочисленных типах синтезов.
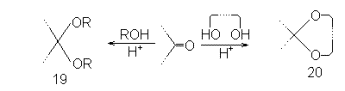
Для того, чтобы циклические производные типа 20 могли образовываться достаточно легко, необходимо соблюдение определенных требований к структуре исходного двухатомного спирта. Две его гидроксильные группы не должны быть расположены слишком далеко одна от другой, так как в противном случае вероятность замыкания цикла резко падает и реакция идет предпочтительно межмолекулярно с образованием линейных олигомеров. Кроме того, возникновение циклической системы не должно вызывать значительных дополнительных напряжений в остальной части молекулы.
По этим причинам возможность образования циклических ацеталей или кеталей подчиняется жесткому контролю со стороны всей структуры, стереохимии и конформации субстрата. В результате реакции, ведущие к таким алкилиденовым производным, протекают весьма избирательно и затрагивают не все, а лишь вполне определенные гидроксильные группы моносахарида или его частично защищенного производного. Таким образом, введение алкилиденовых группировок позволяет резко нарушить монотонность функциональных групп исходных соединений и создает основу для разнообразных способов избирательной защиты спиртовых гидроксилов.