Трансформирующие реакции
Проблема региоспецифичности реакций моносахаридов, как мы видели, может быть разрешена при помощи системы защитных групп. Помимо этого при направленных трансформациях моносахаридов необходимо еще обеспечить их стереоспецифичность, так как в большинстве случаев такие реакции протекают у одного из асимметрических центров и приводят к соединениям, в которых новая группировка также связана с асимметрическим атомом углерода. В целом среди важнейших типов органических реакций наибольшими возможностями, с точки зрения обеспечения стереоспецифичности, обладают ионные реакции, т.е. те, которые протекают через заряженные или высоко поляризованные промежуточные продукты или переходные состояния. Именно такие реакции являются главными инструментами при работе химика-синтетика с углеводами.
Одним из наиболее употребительных типов реакций в этой области являются реакции нуклеофильного замещения при одном из углеродных атомов производного моносахарида. Вот общая схема протекания таких реакций:
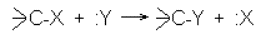
Результатом реакции является обмен заместителей при центральном углеродном атоме. Реагентами служат отрицательно заряженные или нейтральные частицы, имеющие неподеленную пару электронов, так называемые нуклеофилы, такие, например, как HO: -, :CN -, CH 3 COO: -, N 3 -, R 3 N: и т.п. При реакции связь С-Х разрывается таким образом, что уходящая группа Х уносит электронную пару, составляющую ковалентную связь. Для того, чтобы реакция протекала достаточно эффективно, важно, в частности, чтобы Х была, как говорят, хорошо уходящей группой, т.е. имела бы достаточно большое сродство к электрону и образовывала бы достаточно стабильную частицу с неподеленной электронной парой. Хорошие уходящие группы – это те, например, которые при отщеплениии дают высоко стабильные анионы (Cl -, Br -, CH 3 SO 2 O - и т.п.)
Спиртовые гидроксилы – основной тип функции в моносахаридах – по ряду причин являются плохими уходящими группами в нуклеофильном замещении. Поэтому их обычно модифицируют, превращая в эфиры сульфокислот (сульфонаты), чаще всего в эфиры метаносульфокислоты (мезилаты, на схемах обычно обозначаемые знаком Ms) или п-толуолсульфокислоты (тозилаты, обозначаемые обычно Ts). Остаток сульфокислоты – хорошая уходящая группа, легко замещаемая во множестве реакций нуклеофильного замещения, а сам синтез мезилатов и тозилатов выполняется весьма просто, обычно обработкой спирта соответствующим хлорангидридом в пиридине по схеме:
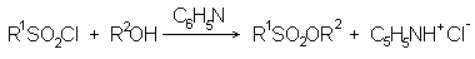
Большое разнообразие нуклеофилов, способных замещать сульфонилоксигруппы в сахарах по механизму S N 2, открывает богатейшие синтетические возможности. Так, например, замещение ацетатом или бензоатом приводит к соответствующим легко омыляемым эфирам, т.е. ведет в конечном счете к обращению конфигурации одного асимметрического центра; замещение на азид с последующим восстановлением позволяет синтезировать аминодезоксисахара; замена на иод или серу ведет к галогендезокси- и к тиосахарам.
Как известно, спирты сравнительно легко окисляются: первичные до карбоновых кислот, а вторичные – до кетонов.
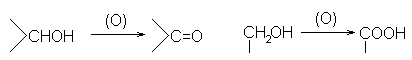
Окисление спиртовых групп в производных моносахаридов по таким схемам широко применяется в синтетической практике. Окисление первично-спиртовой группы служит для синтеза уроновых кислот.
Введение кетогруппы в производные моносахаридов открывает богатейшие синтетические возможности, связанные с чрезвычайно многообразной реакционной способностью карбонильной группы. Основным путем получения таких кетонов служит оксиление производных, содержащих одну вторичную гидроксильную группу. В сравнении с обычными спиртами вторично-спиртовые группы в сахарих поддаются окислению с некоторым трудом. Поэтому для этих целей приходится применять энергичные окислители, такие, как четырехокись рутения или комбинация диметилсульфоксида с реагентами типа ангидридов (уксусный ангидрид, P 2 O 5, дициклогексилкарбодиимид и некоторые другие). Несмотря на некоторую экзотичность этих окислителей, их широко применяют в химии углеводов. Такие методы дают сейчас синтетику возможность окисления практически любой вторично-спиртовой группы и, следовательно, введения карбонильной функции в почти любое желаемое положение.
Восстановительные реакции в химии углеводов употребляются главным образом для введения дезоксизвена и для перехода от карбонильных производных к спиртам, а также для удаления бензильной защиты. Каталитический гидрогенолиз галоген-дезоксисахаров, главным образом иодидов, а также тиопроизводных, ведет к образованию дезоксисахаров по схеме:
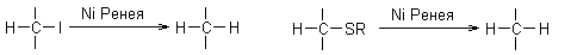
Для синтеза 6-дезоксигексоз обычно применяют ионное восстановление тозилатов алюмогидридом лития (вторичные тозилаты восстанавливаются иначе).
Восстанавливая карбонильную группу комплексными гидридами металлов, например боргидридом натрия, можно получить соответствующие спирты. В циклических кетонах направление подхода реагента и, следовательно, конфигурация образующегося спирта контролируются пространственными факторами, что нередко может обеспечить высокую стереоспецифичность реакции.
Если нужно удлинить цепь углеродных атомов исходного моносахарида или внести в нее разветвления, то возникает необходимость в образовании новых С-С-связей. Наибольшее значение для достижения этих целей имеют реакции карбонильных групп моносахаридов и их производных с реагентами, содержащими нуклеофильный атом углерода, такими, как магнийорганические соединения (например, 87), синильная кислота (88), диазометан (89), илиды (например, 90) и т.п. Общая особенность этих соединений состоит в том, что их можно рассматривать, по крайней мере формально, как доноры карбанионов: частиц с неподеленной электронной парой и отрицательным зарядом на атоме углерода.
Карбонильная группа сильно поляризована: электроны двойной связи оттянуты к атому кислорода, вследствие чего на нем сосредоточен частичный отрицательный заряд, а на атоме углерода – частичный положительный. Поэтому карбонильная группа легко подвергается нуклеофильной атаке, неизменно направленной на ее углеродный атом. В случае соединений с нуклеофильным атомом углерода, как иногда говорят, С-нуклеофилов, такие реагенты приводят к образованию новых С-С-связей.
Конденсация кетопроизводных моносахаридов, содержащих карбонильную группу в циклической системе, может протекать с высокой стереоселективностью, если другие заместители создают значительную неэквивалентность пространственного окружения «верхней» и «нижней» стороны карбонильной группы. В подобных случаях подход реагента осуществляется предпочтительно со стороны наименее экранированной другими группами в молекуле, что и обусловливает образование нового асимметрического центра с определенной конфигурацией. На таких реакциях основаны многие синтезы моносахаридов с разветвленной цепью.
Для синтеза высших сахаров с неразветвленной углеродной цепью применяют сходные реакции С-нуклеофилов с альдегидными группами альдоз. Один из наиболее старых методов в этой области – циангидринный синтез, о котором мы уже говорили в связи с работой Фишера по установлению конфигурации моносахаридов. Использование в подобных реакциях производных моносахаридов с защищенными спиртовыми гидроксилами и свободной альдегидной группой, так называемых аль-форм сахаров значительно расширяет круг применимых реагентов и, следовательно, синтетические возможности реакций. Аналогичное применение находят производные моносахаридов с защищенным гликозидным центром и содержащие альдегидную группу на противоположном конце углеродной цепи.