Газовая хроматография
ГАЗОВАЯ ХРОМАТОГРАФИЯ (ГХ), вид хроматографии, в к-рой подвижной фазой служит газ (пар). В зависимости от агрегатного состояния неподвижной фазы различают газоадсорбционную хроматографию (неподвижная фаза - твердое тело) и газо-жидкостную хроматографию (неподвижная фаза - жидкость, нанесенная тонким слоем на твердый носитель).
Разделение компонентов в газовой хроматографии основано на различии скоростей движения и размывания концентрац. зон исследуемых в-в, движущихся в потоке газовой фазы относительно слоя неподвижной, причем эти в-ва распределены между обеими фазами. Газ-носитель (воздух, N2, Аr, СО2 и др.) должен обычно иметь небольшую вязкость и обеспечивать высокую чувствительность детектирования.
Проведение эксперимента. Газохроматографич. разделение и анализ осуществляются в спец. приборе - газовом хроматографе. В ходе эксперимента газ-носитель из баллона повыш. давления непрерывно поступает в блок подготовки, где дополнительно очищается. Устройство для ввода пробы обычно представляет собой проточную независимо термостатируемую цилиндрич. камеру. Анализируемая проба (1-10мкл) вводится в поток газа при повыш. т-ре дозатором (напр., шприцем) через резиновую термостойкую мембрану. Существуют также автоматич. системы ввода проб (самплеры). Жидкая проба быстро испаряется и потоком газа переносится в хроматографич. колонку, находящуюся в термостате. Разделение обычно проводят при 20-400 °С, но иногда (в осн. при разделении изотопов низкокипящих газов) при значительно более низких т-pax - до т-ры кипения жидкого азота. Для аналит. разделения используют насадочные колонки дл. 0,5-5 м и диам. 0,2-0,6 см, а также капиллярные полые колонки дл. 10-100 м и диам. 0,1-1 мм, и капиллярные насадочные колонки дл. 0,1-20м. Насадкой служат твердый сорбент с развитой пов-стью (50-500 м2/г) или твердый макропористый носитель с уд. пов-стью 0,2-2,0 м2/г, на к-рую тонким слоем нанесена нелетучая жидкость - неподвижная жидкая фаза. Масса жидкой фазы составляет обычно 2-20% от массы носителя. Средний диаметр частиц сорбента 0,1-0,4 мм (колонку заполняют близкими по размеру частицами). Применяют также (обычно в капиллярных наса-дочных колонках) микронасадки с диаметром частиц сорбента 10-50 мкм.
Зоны разделенных компонентов в потоке газа поступают в детекторы хроматографические. В газовой хроматографии используются практически только дифференциальные детекторы (катарометр, пламенно-ионизационный, электронно-захватный, пламенно-фотометрический). Регистратор записывает изменение сигнала во времени. Полученная диаграмма наз. хроматограммой (см. рис.).
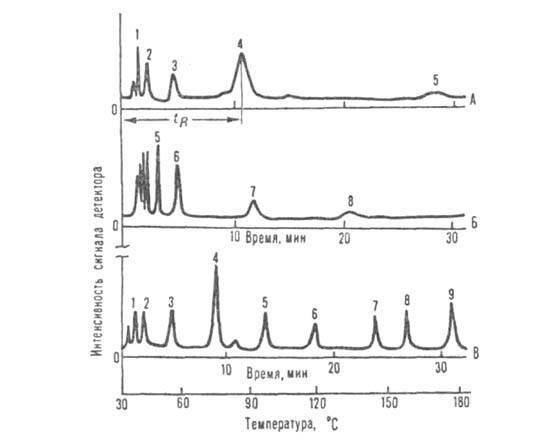
Хроматограммы, полученные при разделении смеси соединений с разл. т-рами кипения: А и Б-изотермич. разделение при 45 и 120°С соотв.; В-разделение при программировании т-ры (скорость повышения т-ры 4,7°С/мин); 1 -пропан; 2-бутан; 3-пентан; 4-гексан; 5-гептан; 6-октан; 7-бромоформ; 8-м-хлортолуол; 9 - броммезити лен.
При использовании сразу неск. детекторов появляется возможность качеств. и количеств. определения состава хроматографич. зон, содержащих два и более соединений. Использование в кач-ве высокоселективного детектора масс-спектрометра привело к созданию высокоэффективного аналит. метода -хромато-масс-спектрометрии. Для управления хроматографом и обработки полученных данных используют ЭВМ. В частности, спец. интеграторы подсчитывают площади пиков на хроматограммах.
Основные измеряемые величины. В газовой хроматографии определяют обычно объем удерживания VR, т.е. объем газа-носителя, прошедший через хроматографич. колонку за время удерживания tR, т.е. время, прошедшее с момента ввода пробы до момента выхода газа с макс. концентрацией определяемого в-ва (напр., на хроматограмме А рисунка показано tR для компонента 4). При этом VR — FctR, где Fc-объемная скорость газа в колонке. Часто определяют также т. наз. исправленный (VR)и относит.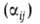 объемы удерживания:
объемы удерживания:
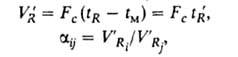
где tм-время удерживания несорбирующегося компонента; tR = tR — tм; V'R. и V'R -соотв. исправленные объемы удерживания в-в i и у. В зависимости от условий эксперимента и диаметра колонки VR может составлять от десятых долей мл до неск. литров.
Для идентификации в-в пользуются относит. объемом удерживания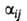 (j-тое в-во - стандартное), а также индексом удерживания Ковача /:
(j-тое в-во - стандартное), а также индексом удерживания Ковача /: 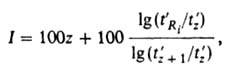
где t'2, t'z+1 и t'Ri.-исправленные времена удерживания н-алканов с числом углеродных атомов z, z + 1 u i-того компонента соответственно; t'Ri. = tRi-tм (tRi-время удерживания i-того компонента). Надежность идентификации по относит. величинам удерживания возрастает при использовании колонок с разными сорбентами.
Эффективность разделения определяется относит. размыванием (расширением) хроматографич. зоны в-ва при движении его вдоль колонки. Ее характеризуют числом N тео-ретич. тарелок (т. т.):
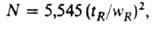
где wR-ширина хроматографич. пика на высоте, соответствующей половине макс. концентрации. Для характеристики колонки широко используют уд. эффективность - число т. т. на 1 м длины колонки (NL)и высоту (H), эквивалентную одной т. т. (ВЭТТ):
где L - длина колонки,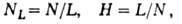 В зависимости от условий эксперимента NL обычно составляет 1000-20000 т.т./м. Зависимость ВЭТТ в насадочной колонке от линейной скорости газа-носителя и приближенно описывается ур-нием Ван-Деемтера:
В зависимости от условий эксперимента NL обычно составляет 1000-20000 т.т./м. Зависимость ВЭТТ в насадочной колонке от линейной скорости газа-носителя и приближенно описывается ур-нием Ван-Деемтера: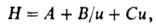 где А и В - коэф. вихревой и продольной диффузии соотв., С - коэф. массо пере дачи.
где А и В - коэф. вихревой и продольной диффузии соотв., С - коэф. массо пере дачи. 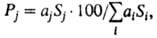
Количественный хроматографич. анализ основан на том, что при постоянных условиях эксперимента интенсивность сигнала детектора прямо пропорциональна концентрации j-того компонента в подвижной фазе, а площадь (Si)соответствующего пика на хроматограмме - его кол-ву. Долю j-того компонента в процентах в n-компонентной смеси рассчитывают по ф-ле где ai и ai- поправочные коэф., зависящие от чувствительности детектора к анализируемым в-вам. Чувствительность анализа определяется обычно чувствительностью детектора; предел обнаружения составляет 10-3-10-6% (при массе пробы 1-10 мг), погрешность 0,2-2%.
Влияние температуры и давления на величину удерживания. Вследствие перепада давления по мере продвижения газа по колонке происходит его расширение и увеличение скорости потока. Истинный объем удерживания VN, рассчитанный с учетом градиента давления по колонке, не зависит от скорости газа-носителя и перепада давления: VN=JV'R, где - т. наз. фактор градиента давления; рi и р0- соотв. давление на входе в колонку и на выходе из нее. Часто рассчитывают уд. объем удерживания Va по ф-ле: 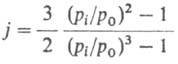
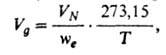
где we-масса неподвижной фазы в колонке; Т - абс. т-ра колонки. Величина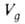 используется для определения ряда физ,-хим. характеристик в соответствии с ур-нием:
используется для определения ряда физ,-хим. характеристик в соответствии с ур-нием:
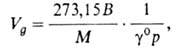
где R - газовая постоянная, М - мол. масса неподвижной жидкой фазы, р - давление насыщ. паров чистого анализируемого соед.,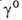 -его коэф. активности. Зависимость
-его коэф. активности. Зависимость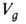 от т-ры описывается ур-нием:
от т-ры описывается ур-нием: 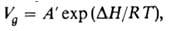
где А' - постоянная,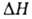 -теплота растворения в-ва в неподвижной жидкой фазе (предполагается, что адсорбционными взаимод. можно пренебречь).
-теплота растворения в-ва в неподвижной жидкой фазе (предполагается, что адсорбционными взаимод. можно пренебречь).
Для разделения смеси соединений, характеризующихся широким интервалом т-р кипения, применяют газовую хроматографию с программированием температуры, когда в процессе хроматографирования в заданные промежутки времени повышают т-ру колонки со скоростью. от неск. °С/мин до неск. десятков °С/мин. Это создает дополнит. возможности расширения области применения газовой хроматографии (сравни хроматограммы на рис.). Для улучшения разделения таких смесей используют также программирование скорости газового потока. При давл. 0,1-2,5 МПа роль газа-носителя сводится в осн. к перемещению исследуемых соед. вдоль колонки. Повышение давления приводит к изменению распределения в-в между подвижной и неподвижной фазами; хроматографич. подвижность многих в-в увеличивается. Газовая хроматография при давлениях газа 10-50 МПа обладает рядом преимуществ по сравнению с жидкостной хроматографией: 1) возможностью целенаправленного изменения объемов удерживания разделяемых соед. путем изменения давления в широких пределах; 2) экспрессностью анализа вследствие меньшей вязкости подвижной фазы и большего значения коэф. диффузии; 3) возможностью использования универсальных высокочувствит. детекторов. Однако сложность аппаратуры и техники работы при повыш. давлении ограничивает широкое распространение этого метода.
Особый интерес представляет хроматографирование с газовой подвижной фазой, находящейся в сверхкритич. состоянии (150-170 °С, давл. до 13,6 МПа). В этих условиях удалось разделить термически нестабильные порфирины. Использование СО2 и NH3 в сверхкритич. состоянии позволило разделить соединения с мол. массой до 40000.
Применение. С помощью газовой хроматографии проводят качеств. и количеств. анализ термически стабильных орг. и неорг. соед., давление пара к-рых при т-ре колонки превышает 0,001 мм рт. ст. (0,13 Па). Газовая хроматография позволяет определять соед., находящиеся в анализируемых пробах в очень малых концентрациях -10-4-10-8%. Широко используется газовая хроматография и для определения разл. физ.-хим. характеристик (констант межфазного распределения, коэф. активности, констант скорости и равновесия хим. р-ций, коэф. диффузии и др.).
Литература
Ж у ховицкий А. А., Ту р кельта у б Н. М., Газовая хроматография, М., 1962; Гольберт К. А., Вигдергауз М.С, Курс газовой хроматографии, 2 изд., М., 1974; Столяров Б. В., Савинов И. М., Витеиберг А. Г., Руководство к практическим работам по газовой хроматографии, 2 изд., Л., 1978; Газовая хроматография с неидеальными элюентами, М., 1980; "Ж. Всес. хим. о-ва им. Д.И. Менделеева", 1983, т 28, № 1. В. Г. Березкин.