Диазосоединeния
ДИАЗОСОЕДИНEНИЯ, содержат группировку N2, связанную с одним орг. остатком. Св-ва алифатич. и ароматич. диазосоединений различаются очень резко.Ароматические диазосоединения ArN2X (Х - остаток к-ты). Связь между ArN2 и X в зависимости от природы последнего м. б. ионной (такие диазосоединения наз. солями диазония, напр.
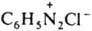 - бензолдиазонийхлорид) или ковалентной, как в соед. ArN=N—X (напр., C6H5N=N—ОН - бензолдиазогидроксид, C6H5N=N—CN - бензолдиазоцианид); соед. ArN—N—ОМ (М - металл) наз. диазотатами металлов. Ковалентные диазосоединения в р-рах диссоциируют с образованием катиона диазония
- бензолдиазонийхлорид) или ковалентной, как в соед. ArN=N—X (напр., C6H5N=N—ОН - бензолдиазогидроксид, C6H5N=N—CN - бензолдиазоцианид); соед. ArN—N—ОМ (М - металл) наз. диазотатами металлов. Ковалентные диазосоединения в р-рах диссоциируют с образованием катиона диазония 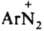 и аниона Х- или превращаются в форму, способную к такой диссоциации (см. ниже). Диазосоединения могут не содержать X, когда Аr имеет отрицат. заряд, как, напр., в случае диазофенолятов (хинондиазидов)
и аниона Х- или превращаются в форму, способную к такой диссоциации (см. ниже). Диазосоединения могут не содержать X, когда Аr имеет отрицат. заряд, как, напр., в случае диазофенолятов (хинондиазидов) 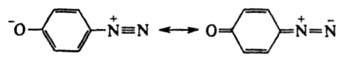
или внутренних солей типа
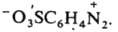 Наиб. важны в практич. отношении соли диазония. Из-за низкой термич. стабильности их обычно используют сразу после получения, не выделяя из р-ров. Твердые соли диазония, у к-рых Х - остаток минер. к-ты (напр., HSO4-, NO3-, Сl-, СlO4-), неустойчивы и часто взрываются. Соли с анионами комплексных к-т (ZnCl3- и BF4-), а также с ArSO3-сравнительно устойчивы. Катион диазония
Наиб. важны в практич. отношении соли диазония. Из-за низкой термич. стабильности их обычно используют сразу после получения, не выделяя из р-ров. Твердые соли диазония, у к-рых Х - остаток минер. к-ты (напр., HSO4-, NO3-, Сl-, СlO4-), неустойчивы и часто взрываются. Соли с анионами комплексных к-т (ZnCl3- и BF4-), а также с ArSO3-сравнительно устойчивы. Катион диазония 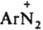 относительно устойчив благодаря сопряжению между ядром и диазониевой группой; последняя оказывает более сильное электроноакцепторное влияние на ароматич. кольцо, чем две нитрогруппы. Два атома N в катионе бензолдиазония линейно расположены в плоскости кольца; расстояние между ними 0,1094 нм. В ИК спектре частота валентного колебания диазониевой группы ионных диазосоединений nN2 в области 2100-2300 см-1 (у ковалентных диазосоединений поглощение отсутствует). В электронном спектре имеются две полосы переноса заряда: 263 нм (lgeA 3,19) и 297 нм (lg el 2,97). Электронодонорные заместители в орто- и пара-положениях бензольного кольца оказывают батохромное влияние на спектральные характеристики (см. Цветность органических соединений) и повышают термостабильность катиона.
относительно устойчив благодаря сопряжению между ядром и диазониевой группой; последняя оказывает более сильное электроноакцепторное влияние на ароматич. кольцо, чем две нитрогруппы. Два атома N в катионе бензолдиазония линейно расположены в плоскости кольца; расстояние между ними 0,1094 нм. В ИК спектре частота валентного колебания диазониевой группы ионных диазосоединений nN2 в области 2100-2300 см-1 (у ковалентных диазосоединений поглощение отсутствует). В электронном спектре имеются две полосы переноса заряда: 263 нм (lgeA 3,19) и 297 нм (lg el 2,97). Электронодонорные заместители в орто- и пара-положениях бензольного кольца оказывают батохромное влияние на спектральные характеристики (см. Цветность органических соединений) и повышают термостабильность катиона. 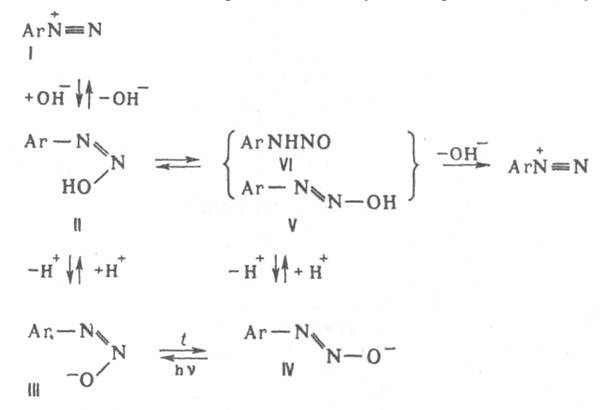
При взаимод. катиона диазония (I) с ОН- образуется цис-диазогидроксид (II) - амфотерное соед., диссоциирующее с образованием циc-диазотата (III). Константа кислотности I ниже, чем II, поэтому равновесная концентрация последнее мала. Равновесие между I и III устанавливается за несколько мс. Щелочная соль цис-диазотата в твердом виде крайне неустойчива. В щелочном р-ре при нагр. циc-диазотат превращается в транс-форму (IV), щелочные соли к-рой стабильны; они бесцветны или окрашены в желтый цвет Обратному переходу IV в III способствует УФ облучение. При подкислении бифункциональный анион IV превращается в транс-диазогидроксид (V) или N-нитрозамин (VI), к-рые далее переходят в катион I. Из транс-диазотата (IV) катион диазония I образуется относительно медленно, т.к. они не находятся в состоянии простого протолитич. равновесия. Наличие в нейтральных и слабокислых средах N-нитрозамина способствует частичному дедиазотированию и образованию амина ArNH2. Если X = SO23- или CN-, то образуются ковалентные цис- и транс-диазосульфонаты Аr—N=N—SO3Na либо диазоцианиды Ar—N=N—CN. Однако эти транс-изомеры, в отличие от транс-диазогидроксида (V), в темновых условиях в катион I не переходят, а поэтому и не способны к азосочетанию. Диазониевая группа сильно активирует нуклеоф. замещение; так, в 2,4-динитробензолдиазонии в нейтральной среде одна из NO2-групп, преим. в положении 2, обменивается на группу ОН. К р-циям, идущим с превращением диазогруппы, относится замена ее основанием при нагр. диазосоединений в разб. H2SO4 (типичная р-ция SN1:
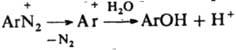 ). При недостаточной кислотности среды образуются т. н. диазосмолы, что часто является причиной низкого выхода и плохого качества фенолов и азокрасителей, получаемых в соответствующих произ-вах. Аналогично, по гетеролитич. механизму в водном р-ре спирта диазогруппа замещается на алкокси-группу RO. При восстановлении диазосоединений абс. спиртом выделяется N2 и образуется неустойчивый арильный радикал, отрывающий Н от спирта. Восстановителями могут служить также Н3РО3, гидрохинон и др. Арильные радикалы, образующиеся при электрохим. восстановлении солей диазония, а также при восстановлении порошкообразной Сu, частично рекомбинируют с образованием симметричных биарилов, увеличению выхода к-рых способствует наличие oртo-заместителей (см. Гаттермана синтез). Соли диазония в присут. солей Cu(I) и нек-рых др. металлов легко замещают диазо-группу атомом галогена, а также группами CN, NCS, NO2, HS, RS, SO2H и др. (см. Зандмейера реакция). По свободно-радикальному механизму под действием водного р-ра щелочи разлагаются также ковалентные диазосоединения, напр., диазогидроксид; в присут. ароматич. углеводорода в результате арилирования последнего образуются несимметричные углеводороды (р-ция Гомберга). В кислых средах при действии SnCl2 соли диазония восстанавливаются в арилгидразины:
). При недостаточной кислотности среды образуются т. н. диазосмолы, что часто является причиной низкого выхода и плохого качества фенолов и азокрасителей, получаемых в соответствующих произ-вах. Аналогично, по гетеролитич. механизму в водном р-ре спирта диазогруппа замещается на алкокси-группу RO. При восстановлении диазосоединений абс. спиртом выделяется N2 и образуется неустойчивый арильный радикал, отрывающий Н от спирта. Восстановителями могут служить также Н3РО3, гидрохинон и др. Арильные радикалы, образующиеся при электрохим. восстановлении солей диазония, а также при восстановлении порошкообразной Сu, частично рекомбинируют с образованием симметричных биарилов, увеличению выхода к-рых способствует наличие oртo-заместителей (см. Гаттермана синтез). Соли диазония в присут. солей Cu(I) и нек-рых др. металлов легко замещают диазо-группу атомом галогена, а также группами CN, NCS, NO2, HS, RS, SO2H и др. (см. Зандмейера реакция). По свободно-радикальному механизму под действием водного р-ра щелочи разлагаются также ковалентные диазосоединения, напр., диазогидроксид; в присут. ароматич. углеводорода в результате арилирования последнего образуются несимметричные углеводороды (р-ция Гомберга). В кислых средах при действии SnCl2 соли диазония восстанавливаются в арилгидразины: 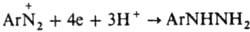 . Под действием света соли диазония разлагаются, причем особенно легко, если в орто-и пара-положениях находятся сильные электронодонорные заместители; в результате фотолиза выделяется N2, происходит сужение кольца и образуются высокомол. соединения. В пром-сти соли диазония получают диазотированием ароматич. аминов. Диазосоединения можно синтезировать также действием на фенол HNO2, взятой в избытке (промежуточно образуется нитрозофенол):
. Под действием света соли диазония разлагаются, причем особенно легко, если в орто-и пара-положениях находятся сильные электронодонорные заместители; в результате фотолиза выделяется N2, происходит сужение кольца и образуются высокомол. соединения. В пром-сти соли диазония получают диазотированием ароматич. аминов. Диазосоединения можно синтезировать также действием на фенол HNO2, взятой в избытке (промежуточно образуется нитрозофенол): С6Н5ОН + 4HNO2 : -ОС6Н4N2+ + 2HNO3 + 2Н2О
Диазотаты образуются по р-ции N-нитрозоациларилидов в щелочной среде:
ArN(NO)COCH3 + 2КОН : ArN2OK + СН3СООК + Н2О
В основе большинства методов определения диазосоединений лежит азосочетание. Если диазосоединение находится в форме транс-диазотата, его предварительно нужно действием к-ты перевести в активную диазониевую форму. Ионные и ковалентные диазосоединения различают по наличию у первых в ИК спектре частоты валентного колебания связи N=N. В пром-сти для определения диазосоединений используют автоматизир. контроль, основанный на электрохим. измерениях. Ароматич. диазосоединения применяют преим. для получения азокрасителей, а также как светочувствит. материалы для изготовления фоторезистов и в диазотипии (см. Репрография): в пром. орг. синтезе - для получения ценных промежуточных продуктов. Ароматич. диазосоединения открыты П. Гриссом в 1858.
Алифатические диазосоединения (диазоалканы) RR'CN2 (R,R' = Н, Alk). Эти соед. окрашены в цвета от желтого до пурпурно-красного, ядовиты. Низшие - взрывоопасные газы, высшие - жидкие или твердые в-ва, более устойчивы. Алифатич. диазосоединения стабилизируются в форме илида:
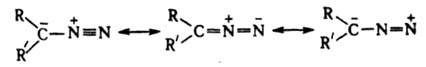
Длина связи N-N в диазометане 0,113 нм, что ближе к длине тройной, чем двойной связи. Частота валентного колебания nN лежит в области 2000-2200 см-1. Диазоалканы способны присоединять протон и к-ты Льюиса, давая соли диазония, к-рые легко разлагаются с выделением N2 и образованием продуктов превращения алкильного катиона. Вступая в р-цию 1,3-биполярного присоединения с алкенами, диазоалканы дают производные пиразолина, при фотолизе из алифатич. диазосоединений образуются карбены. Получают диазоалканы след. методами: 1) щелочной обработкой ацил-, карбамоил- или тозил-N-нитрозаминов (см. Диазометан), а также N-тозилгидразонов, напр.:
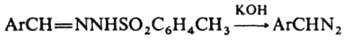
2) окислением гидразонов:
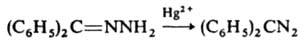
3) обработкой оксимов хлорамином или О-сульфонилгидроксиламином (HO3SONH2):
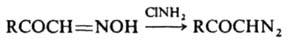
4) действием HNO2 на a-аминоэфиры:
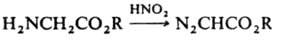
Диазоалканы - алкилирующие агенты. Литература