Реакции в растворах
РЕАКЦИИ В РАСТВОРАХ, хим. р-ции, идущие между растворенными в-вами, а также между растворенным в-вом и р-рителем. Как правило, р-ритель принимает непосредств. участие в хим. взаимодействии. С одной стороны, р-ритель проявляет себя как сплошная среда (континуум), характеризующаяся вязкостью, диэлектрич. проницаемостью, поляризацией и т. п.; с другой стороны-как совокупность молекул, обладающих размером, дипольным моментом, поляризуемостью и участвующих в межмолекулярном взаимодействии друг с другом и с молекулами растворенного в-ва, к-рое м. б. описано соответствующими потенциалами. Молекулы р-ри-теля образуют упорядоченные структуры на расстояниях, сравнимых с межмолекулярными (ближний порядок), что отражается видом корреляц. ф-ции распределения (см. Жидкость). Сольватные оболочки, окружающие реагирующие частицы в р-ре, характеризуются координац. числом и ре-лаксац. параметрами.
Термодинамика. Если при переходе реагентов А и В из газовой фазы в р-р не происходит изменения в направлении р-ции А + В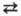 С и составе продуктов, положение равновесия реакций в растворах зависит от своб. энергии сольватации р-рителем
С и составе продуктов, положение равновесия реакций в растворах зависит от своб. энергии сольватации р-рителем 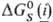 исходных реагентов и продуктов. Стандартная энергия Гиббса реакций в растворах
исходных реагентов и продуктов. Стандартная энергия Гиббса реакций в растворах 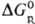 связана со стандартной энергией Гиббса соответствующей газофазной р-ции
связана со стандартной энергией Гиббса соответствующей газофазной р-ции 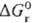 соотношением:
соотношением: 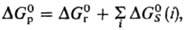 где
где 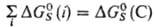
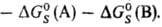
Величина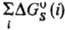 м. б. очень значительной, сравнимой с
м. б. очень значительной, сравнимой с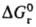 или даже превышающей ее. Напр., в случае ионов в полярных р-рителях
или даже превышающей ее. Напр., в случае ионов в полярных р-рителях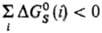 и если
и если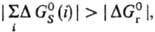 в р-ре энергетически выгодными становятся процессы ионизации, к-рые в газовой фазе практически не идут.
в р-ре энергетически выгодными становятся процессы ионизации, к-рые в газовой фазе практически не идут.
Кинетика. Важнейшей особенностью реакций в растворах является многообразие равновесных форм, в к-рых исходный реагент может участвовать в р-ции. В зависимости от природы р-рителя и реагента в р-ре может происходить, с одной стороны, ионизация молекул растворенного в-ва с образованием ионных пар (контактных и сольватно разделенных) и дальнейшей их диссоциацией на ионы, с другой - образование гомо- и гетероассоциатов и кластеров с участием молекул как реагентов, так и р-рителя. Последнее характерно для молекул, образующих водородные связи (Н-связи).
Наблюдаемые константы скорости реакций в растворах, за редким исключением, являются эффективными величинами. Простые порядки р-ции, полученные при обработке данных кинетич. эксперимента, проведенного в узком интервале т-р и концентраций, как правило, являются кажущимися и не отражают истинной молекулярности р-ции, то же откосится и к энергии активации р-ции. Поэтому, прежде чем обсуждать вопрос о влиянии р-рителя на константу скорости реакции в растворах, надо установить, какая из форм исходного реагента принимает участие в лимитирующей стадии сложной р-ции. Это же относится к квантовохим. расчетам, в основу к-рых следует закладывать строение частиц не исходных реагентов, а тех форм, в к-рых эти соед. реагируют в р-ре.
При наличии в системе разл. активных форм исходных реагентов р-ция идет по неск. каналам. Наблюдаемая константа скорости является ф-цией констант скорости р-ций в каналах и соотношений между концентрациями разл. форм. Напр., если в-во М:А, участвующее в р-ции 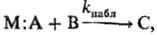 образует в р-ре ионные пары и своб. ионы, в р-ции могут участвовать все формы, находящиеся в равновесии: молекулярная М:А, контактные ионные пары М + · А- , сольватно разделенные ионные пары М+|S|А-(S-p-ритель), своб. ионы М++А-; каждой из этих форм соответствует своя константа скорости ki. Наблюдаемая константа скорости
образует в р-ре ионные пары и своб. ионы, в р-ции могут участвовать все формы, находящиеся в равновесии: молекулярная М:А, контактные ионные пары М + · А- , сольватно разделенные ионные пары М+|S|А-(S-p-ритель), своб. ионы М++А-; каждой из этих форм соответствует своя константа скорости ki. Наблюдаемая константа скорости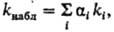 где a(i)-доля i-й формы.
где a(i)-доля i-й формы.
В случае, когда присутствуют только две формы, напр. М:А и М +·А-, реагирующие с В в двух простых параллельных стадиях с константами скорости k1 и k2 соотв., kнабл = ak1+ +(1—a)k2. Если константа равновесия перехода между двумя формами К=(1—a)/a, то вышеприведенное выражение принимает вид: kнабл=(k1 + Kk2)/(1+К), Обычно реакц. способность ионной пары выше, чем молекулы, и в случае, когда Kk2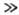 k1, выражение упрощается: kнабл= = Kk2/(1 + К).
k1, выражение упрощается: kнабл= = Kk2/(1 + К).
Для р-ции типа X+Y: продукты, где X и Y-соед., способные образовывать Н-связи: вода, спирты, амины, карбоновые к-ты и т. п., характерно участие гомоассоциатов Хn и Ym, а также гетероассоциатов XpYq. Во мн. случаях р-ции между мономерными формами X и Y практически отсутствуют. Имеют место след. равновесия:
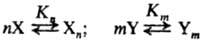
(n = 1, 2, ...,р, m= 1, 2, ..., q)
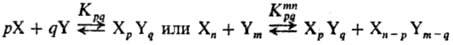
Благодаря ассоциации удлиняются хим. связи, увеличиваются эффективные электрич. заряды на атомах реагентов. Квантовохим. расчет показывает, напр., что в результате димеризации метанола связь О—Н удлиняется на 4%, заряд-на атоме Н увеличивается на 25% и на атоме О-на 14%. Дальнейшая ассоциация приводит к еще большему росту зарядов. Указанные изменения в структуре молекулы, как правило, приводят к увеличению реакц. способности спирта. Помимо этого, меняется сам механизм р-ции в связи с возможностью образования циклич. предреакц. комплексов и циклич. переходных состояний. При этом уменьшаются стерич. затруднения и снижается энергетич. барьер при осуществлении элементарного акта р-ции: разрыв и образование связей в циклич. "заготовке" происходят согласованно (см. Согласованные реакции). Напр., в р-ции ROH + + R'NCO : R'NHCOOR мономер спирта практически не-реакционноспособен и р-ция идет только с участием диме-ров и тетрамеров спирта:
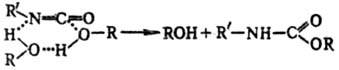
Существенно, что наряду с активными гетероассоциатами XpYq, оптимальными по составу и структуре ("удачными микрореакторами"), через к-рые осуществляется образование продукта (канал выхода реакции), в р-ре возникает большое число разнообразных неактивных (тупиковых) ас-социатов XiYj, в виде к-рых находится осн. масса реагентов ("канал накопления"). Схема реакции в растворах с участием ассо-циатов выглядит след. образом:
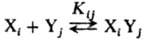 (канал накопления)
(канал накопления)
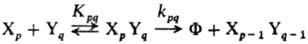 (канал выхода)
(канал выхода)
Согласно схеме, при наличии одного канала выхода скорость образования продукта Ф пропорциональна концентрации реакционноспособного ассоциата [ХрYq]: w = = d[Ф]/dt = kpq[ХрYq]; эта концентрация является ф-цией констант равновесия Кp, Кq, Кpq, Кi, Kj, Кij и стехиометрич. коэффициентов i,j,p,q. Наблюдаемый порядок реакции в растворах в этом случае зависит от соотношения стехиометрич. коэф., с к-рыми входят реагенты в каналы накопления и выхода. Энергия активации р-ции и предэкспоненц. множитель, формально найденные из аррениусовской температурной зависимости наблюдаемой константы скорости, являются сложными величинами, включающими термодинамич. характеристики равновесий образования вышеуказанных гомо- и гетероассоциатов.
Роль диффузии. В отличие от газовой фазы, реагирующие частицы в р-ре окружены молекулами р-рителя и не могут претерпевать двойных соударений. Сближение частиц А и В, необходимое для осуществления р-ции А + В : продукты, реализуется путем взаимной диффузии с относит. коэффициентом DАВ=DА+DВ, где DА и DВ-коэф. диффузии А и В в р-ре. Число "первых встреч", в результате к-рых образуется "диффузионная пара"-две расположенные рядом незаряженные частицы, окруженные молекулами р-рителя ("клеткой"),-рассчитывается по ф-ле Смолуховского: ZB = 4pDАВ(rА + rB) [см3/(молекул·с)], где rА и rB-радиусы А и В. Для близких по размеру частиц (rА ! rB) ZB = = 8 RT/3·103h [л/(моль·с)] (T-абс. т-ра, R-газовая постоянная). Здесь ZB-число первых встреч за 1 с при единичных концентрациях А и В, h-вязкость среды. Как видно из ур-ния, ZB обратно пропорционально вязкости.
Если скорость р-ции между А и В очень велика (рекомбинация своб. радикалов или разнозаряженных ионов, электронные и протонные переходы), т.е. р-ция происходит уже при первой встрече, скорость ее лимитируется диффузией (см. Диффузионно-контролируемые реакции). Константа скорости такой р-ции kдиф ! ZB и также обратно пропорциональна вязкости. В случае обычных молекулярных и ионных р-ций (энергия активации Е > 40 кДж/моль) реагенты А и В, попав в "клетку" р-рителя, претерпевают множественные соударения, число к-рых пкл тем больше, чем дольше они там находятся, т. е. чем больше вязкость среды (пкл ~ h). Время пребывания частицы в клетке т (среднее "время оседлой жизни", по Я. И. Френкелю) м. б. найдено по ур-нию: т=т0exp(Eклl/RT), где т0!10-13-10-12 с-период колебаний частиц в клетке, Екл энергия, необходимая для выхода частицы из клетки. Для воды т0 = 1,4·10~12 с, т= 1,7·10-9с, Eкл!7RT, т.е. при 300 К Eкл! 17кДж/моль. Величина пкл ! т/т0 = ехр(Eкл/RT), что для большинства жидкостей составляет 102 — 104.
Полное число соударений Z = ZBпкл, т.е. равно произведению двух независимых вероятностей, одна из к-рых прямо пропорциональна, другая - обратно пропорциональна вязкости. Поэтому константа скорости обычной р-ции не зависит от вязкости р-рителя, вплоть до стеклообразного состояния.
Если собственно хим. превращение в диффузионной паре (константа скорости kкл) происходит настолько медленно, что необходимо учитывать не только константу скорости образования пары k1 = kдиф, но и константу скорости се распада k2, то реакцию в растворе можно представить схемой:
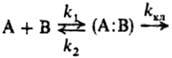 продукты
продукты
Диффузионная пара (А: В) обычно является статистич. образованием ("столкновительным комплексом", по Я. Бьерру-му). Однако при наличии между А и В межмол. взаимод. пара (А:В) м.б. комплексом (напр., донорно-акцепторным) или ассоциатом, в состав к-рого могут входить и молекулы р-рителя. По сути, упоминавшиеся выше гетероассоциаты-это сложные диффузионные пары. Согласно этой схеме, реакции в растворах-двустадийный процесс, в к-ром первая стадия диффузионная, а вторая - химическая, т е. бимолекулярная
Реакция в растворе сводится к мономолекулярному превращению диффузионной пары. В стационарном состоянии реагирующей системы наблюдаемая константа скорости в ур-нии d [продукт]/dt = kнабл [А]·[В] имеет вид: kнабл = k1·kкл/(k2+kкл). Если хим. р-ция в клетке медленнее, чем диффузия (k2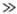 kкл), выражение для kнаб упрощается: kнабл = KАВkкл, где КАВ = = k1/k2, по сути, есть константа равновесия образования диффузионной пары.
kкл), выражение для kнаб упрощается: kнабл = KАВkкл, где КАВ = = k1/k2, по сути, есть константа равновесия образования диффузионной пары.
Теория активированного комплекса используется для расчета истинной константы скорости элементарной реакции в растворе. Предполагается, что точно определена форма реагента (хим. субстрат), превращение к-рого приводит к образованию продуктов, причем лимитирующей стадией является не диффузия молекул в клетку р-рителя, а собственно превращение субстрата внутри клетки (кинетич. режим протекания р-ции). Для реакций в растворах, к-рые протекают при постоянном давлении, осн. соотношение теории связывает константу скорости р-ции k со своб. энергией активации DG. - изменением энергии Гиббса системы при переходе от реагентов к переходному состоянию (ПС):
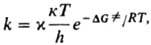
где Т-.абс. т-ра, к и h-постоянные Больцмана и Планка соотв., х - трансмиссионный множитель, к-рый в рамках теории активир. комплекса не м. б. вычислен и считается равным 1. Величину DG. можно представить в виде суммы двух слагаемых, одно из к-рых (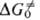 ) учитывает изменение своб. энергии изолир. субстрата, второе (
) учитывает изменение своб. энергии изолир. субстрата, второе (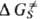 )-эффекты сольватации:
)-эффекты сольватации:
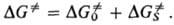
Последние особенно существенны при сильном электроста-тич. взаимод. между субстратом и средой при условии, что распределения электрич. заряда в субстрате и в ПС значительно различаются, т.е. р-ция является процессом с переносом заряда. Напр., в простейших р-циях нуклеоф. замещения SN2:
Х- +СН3Y:[Х...СН3..Y]-:XCH3 + Y-
(X, Y-Hal, ОН и т.п., Х-, Y--соответствующие анионы), отрицат. заряд в реагентах локализован на частице X, а в ПС делокализован. ПС сольватировано слабее реагентов, т.е. 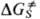 положит. величина. В полярных р-рителях константа скорости р-ций типа SN2 уменьшается на 10-20 порядков по сравнению со значением в газовой фазе. Существ. вклад в величину
положит. величина. В полярных р-рителях константа скорости р-ций типа SN2 уменьшается на 10-20 порядков по сравнению со значением в газовой фазе. Существ. вклад в величину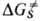 могут давать также изменения прочности водородных связей субстрата и среды. Для р-ций неполярных молекул и для р-ций в неполярных апротонных р-рителях, когда электростатич. взаимод. слабы и прочные водородные связи не образуются, истинные константы скорости, как правило, мало отличаются от газофазных значений.
могут давать также изменения прочности водородных связей субстрата и среды. Для р-ций неполярных молекул и для р-ций в неполярных апротонных р-рителях, когда электростатич. взаимод. слабы и прочные водородные связи не образуются, истинные константы скорости, как правило, мало отличаются от газофазных значений.
Для газофазных р-ций аррениусовскую энергию активации отождествляют с высотой барьера на поверхности потенциальной энергии (ППЭ), а слабо зависящий от т-ры предэкспоненц. множитель выражают через вклады реагентов и ПС в энтропию системы. Своеобразие реакций в растворах состоит в том, что в ППЭ в качестве независимых переменных наряду с координатами хим. субстрата xi (длинами связей, валентными углами и т. п.) введены координаты среды - изменения положения и ориентации молекул р-рителя относительно молекул субстрата и друг друга. При этом существует огромное кол-во конфигураций сольватной оболочки хим. субстрата, соответствующих разл. локальным минимумам ППЭ. Эти минимумы близки по энергии и разделены невысокими (несколько кДж/моль) потенц. барьерами. В такой ситуации к.-л. один определенный минимум теряет значение и рассматривают усредненное влияние всех конфигураций сольватной оболочки на энергетику хим. превращения субстрата. Усредненная таким образом ППЭ представляет собой поверхность свободной энергии (ПСЭ) G(xi), зависящую лишь от координат хим. субстрата. Изменение своб. энергии DG. отождествляют с высотой барьера на ПСЭ, разделяющего минимумы реагентов и продуктов. При вычислении ПСЭ учитывают взаимод. молекул р-рителя между собой и с молекулами субстрата; при сильном взаимод. ПСЭ существенно отличается от газофазной ППЭ. Сольватация меняет не только высоту барьера, но и структуру субстрата в стационарных точках (реагенты, ПС), поэтому вычисление сольватац. вклада 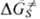 не сводится к простому расчету своб. энергии сольватации для соответствующих газофазных структур.
не сводится к простому расчету своб. энергии сольватации для соответствующих газофазных структур.
Прямое вычисление энергетич. профиля вдоль координаты р-ции на ПСЭ и последующий расчет константы скорости возможны с применением методов Монте-Карло и мол. динамики (см. Молекулярная динамика). Для упоминаемой р-ции SN2 при X, Y = С1 в газовой фазе ППЭ, полученная квантовохим. расчетом, имеет сложный профиль с двумя ямами, соответствующими образованию пред-и послереакционного комплексов. Для той же р-ции в воде профиль ПСЭ, полученный сочетанием квантовохим. расчета с методом Монте-Карло, имеет обычный вид кривой с потенц. барьером (см. рис.). Кол-во молекул р-рителя, к-рые следует учесть при расчете ПСЭ, велико. Как минимум, это молекулы, входящие в неск. ближайших коорди-нац. сфер хим. субстрата. Для р-ции SN2 в воде, напр., явно учитывали 250 молекул Н2О; полученное значение DG. = = 110 кДж/моль при 25 °С хорошо согласуется с экспериментом. Наконец, применение методов мол. динамики позволяет выйти за рамки теории активир. комплекса, рассчитав трансмиссионный множитель х в выражении для константы скорости.
Энергетич. профиль пов-сти потенц. энергии для р-ции SN2 в газовой фазе (пунктирная линия) и пов-сти своб энергии для р-ции в р-ре (сплошная линия).
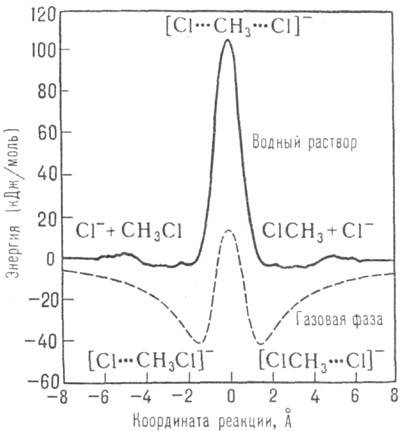
Континуальная модель растворителя. Высоту барьера ПСЭ можно определить с меньшими вычислит. затратами на основе континуальной модели р-рителя. Обычно этот подход применяют к р-циям с переносом заряда в апротонных полярных средах, когда взаимод. среды и субстрата в осн. электростатическое. Р-ритель рассматривают как сплошной диэлектрич. континуум с диэлектрич. проницаемостью es. В нем вырезают полость, в к-рую помещен реагирующий хим. субстрат (диэлектрич. проницаемость полости равна 1). Зарядовое распределение субстрата поляризует среду; поле наведенной поляризации среды, в свою очередь, меняет зарядовое распределение субстрата. Результирующая поляризация среды Р(r) в точке пространства вне полости вместе с результирующим зарядовым распределением субстрата r(r) рассчитывается методом итераций. На каждой итерации электростатич. расчет Р(r) комбинируют с квантовохим. вычислением r(r). Этот метод расчета электростатич. составляющей сольватации наз. методом самосогласованного реакционного поля (метод ССРП). В простейших реализациях, если моделировать хим. субстрат в виде помещенных в центр сферич. полости точечных иона и диполя, метод позволяет получить для энергии сольватации известные ф-лы Борна и Онсагера (см. Сольватация):
Динамическая континуальная модель и коллективные координаты среды. При наличии сильного взаимод. субстрата со средой необходимо в явном виде учесть зависимость ПСЭ и своб. энергии активации DG. от изменений, происходящих в среде. Континуальная модель вводит в качестве переменных среды не элементарные смещения отдельных молекул р-рителя, а коллективные координаты (моды), описывающие согласованные комбинации большого числа элементарных смещений. Такой подход в принципе позволяет выделить одну или неск. существенных для описания р-ции коллективных координат (кинетически активных мод) и ограничиться их явным рассмотрением.
При чисто электростатич. взаимод. среды и субстрата в качестве коллективной координаты среды принимают т. наз. инерционную поляризацию Pin(r), т.е. часть полной поляризации среды, из к-рой исключена поляризация электронных оболочек молекул р-рителя (С. И. Пекар, 1951; Р. Маркус, 1956). Инерционная поляризация изменяется со временем независимо в каждой точке r трехмерного пространства, т.е. ее значения в каждой точке-независимые динамич. переменные. Запись Pin(r) означает, что рассматривается бесконечный набор (континуум) всех таких переменных. При заданном зарядовом распределении субстрата rа(r) ПСЭ представляет собой бесконечномерный параболоид в пространстве поляризаций. Его минимум соответствует поляризации 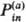 (r), равновесно подстроенной под заряд rа(r). При др. зарядовом распределении rb(r) минимум параболоида ПСЭ смещается в др. точку пространства поляризации
(r), равновесно подстроенной под заряд rа(r). При др. зарядовом распределении rb(r) минимум параболоида ПСЭ смещается в др. точку пространства поляризации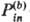 (r). Если в ходе р-ции распределение заряда субстрата меняется от rа(r) до rb(r), ПСЭ комбинируется из двух пересекающихся параболоидов. Такая ПСЭ описывает реорганизацию полярного р-рителя при учете одних лишь электростатич. сил.
(r). Если в ходе р-ции распределение заряда субстрата меняется от rа(r) до rb(r), ПСЭ комбинируется из двух пересекающихся параболоидов. Такая ПСЭ описывает реорганизацию полярного р-рителя при учете одних лишь электростатич. сил.
Переходное состояние для р-ции переноса электрона характеризуется высотой барьера DG., к-рая выражается уравнением Маркуса:
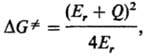
где Q-разность своб. энергий системы продуктов и системы реагентов, т.е. разность между :положениями минимумов соответствующих параболоидов на энергетич. шкале (для экзотермич. р-ций Q < О), а Еr-т. наз. энергия реорганизации р-рителя. Этот важный параметр теории характеризует степень изменения ПСЭ при изменении распределения заряда на субстрате от rа(r) до rb(r). Скорость изменения поляризации среды, подстраивающейся под зарядовое распределение субстрата, к-рое меняется в ходе р-ции, определяется диэлектрич. релаксацией р-рителя. Время продольной диэлектрич. релаксации тL является динамич. характеристикой среды, оно м. б. определено экспериментально на основе ф-лы Дебая (см. Диэлектрики).
Стохастическая теория. В динамич. теории подлежит рассмотрению огромное кол-во координат среды. Эффективный способ обойти эту трудность-рассматривать в явном виде только координаты хим. субстрата, к-рые, однако, взаимодействуют (обмениваются энергией) с неучтенными в явном виде переменными среды, т.е. образуют термодинамически открытую систему. Динамика открытых систем описывается стохастич. ур-ниями, простейшее из к-рых-уравнение Ланжевена:
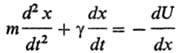 + случайная сила,
+ случайная сила,
где x-динамич. переменная (обобщенная координата), m-связанная с ней масса. Первыми в правой и левой частях этого ур-ния стоят обычные ньютоновские члены: кинематич. член, пропорциональный ускорению, и регулярная сила [U(x)-потенц. энергия]. Второй член в левой части-сила трения, к-рая пропорциональна скорости dx/dt (g-коэф. трения); она описывает диссипацию энергии вследствие взаимод. координаты х со средой. Второй член в правой части-случайная сила-описывает нерегулярное воздействие среды на переменную х (при моделировании реакций в растворах она обусловливает термич. активацию субстрата, необходимую для преодоления потенц. барьера). Сила трения и случайная сила связаны таким образом, что их комбинир. воздействие обеспечивает установление в системе больцмановского распределения спустя время, превышающее период релаксации переменной х.
Впервые ур-ние Ланжевена было предложено для объяснения броуновского движения взвешенных в жидкости коллоидных (т.е. макроскопич.) частиц. В теории реакций в растворах координата х обычно соответствует одной из степеней свободы хим. субстрата, т.е. является микроскопич. переменной. Поэтому интерпретация коэф. у как макроскопич. трения, обусловленного вязкостью среды, неправомерна, хотя ее иногда и используют (см. ниже). Величину у следует рассматривать как феноменология, характеристику взаимод. между субстратом и средой, ее микроскопич. интерпретация требует спец. теории.
Если U(х)-потенц. кривая с барьером, ур-ние Ланжевена описывает динамику перехода системы через барьер и м. б. использовано для описания кинетики реакций в растворах. Приближенная теория, разработанная Г. Крамерсом (1940), дает для константы скорости k аррениусовскую зависимость от т-ры, причем энергия активации Е совпадает с высотой барьера, а предэкспоненц. множитель зависит от g. При больших g эта зависимость обратно пропорциональна. Иногда полагают, что у пропорциональна гидростатич. вязкости среды h; в таком случае расчет предсказывает зависимость k ~ 1/h) (предельный случай большой вязкости). Эксперим. данные в отдельных случаях подтверждают этот вывод. Следует помнить, что речь идет о влиянии вязкости среды на элементарный акт хим. р-ции, а не о тривиальном влиянии, к-рое всегда наблюдается, если р-ция протекает в диффузионном режиме, как было рассмотрено в соответствующем разделе данной статьи.
Перенос электрона. Для мн. р-ций, в к-рых донор электрона Д и акцептор А представляют собой комплексы переходных металлов, орг. субстраты или фрагменты биол. молекул, перенос электрона предполагается внешнесферным. Это означает, что на лимитирующей стадии р-ции внутр. координаты субстрата не меняются. Меняется только инерционная поляризация среды Pin(r) в соответствии с изменением зарядового распределения r(r). Перенос электрона м.б. адиабатическим и неадиабатическим. В первом случае рассматривается лишь изменение в ходе р-ции единственного электронного состояния (основного или возбужденного), взаимодействующей пары ДА, во втором-по крайней мере двух состояний. В случае адиабатич. переноса электрона можно пользоваться ур-нием Ланжевена, причем переменная х выражается через Pin(r), т. е. оказывается коллективной модой среды, описывающей ее реорганизацию, а коэф. трения у пропорционален времени продольной релаксации тL, т.е. предэкспоненц. множитель А ~ 1/тL. Такая зависимость экспериментально наблюдается для нек-рых р-ций (см. Реакционная способность). Иногда имеет место более слабая зависимость А ~ (1/тL)b, где b < 1, к-рую интерпретируют усложнением модели р-ции и соответствующего стохастич. ур-ния. Динамика неадиабатич. переноса электрона впервые изучалась В. Г. Левичем и Р. Р. Догонадзе (1959). В их теории реакции в растворах рассматривается как безызлучат. квантовый переход, причем зависимость константы скорости от релаксац. характеристик р-рителя отсутствует.
Неравновесная сольватация. Говоря о неравновесной сольватации, обычно имеют в виду, что координата р-ции х описывает движение ядер в субстрате (а не в среде, как при переносе электрона), и в ходе элементарного акта р-ции координаты р-рителя qv не успевают подстраиваться под движение системы по х, к-рое считается более быстрым. В предельно неравновесном случае конфигурация сольват-ной оболочки субстрата в ПС остается такой же, какой она была в конфигурации реагентов (предельный случай "замороженной" среды).
Неравновесные кинетич. режимы реакций в растворах возникают, когда сильно различаются характеристич. времена, связанные с движениями в хим. подсистеме (тx) и в среде (тq). Из сказанного выше очевидно, что тq следует относить именно к коллективным модам среды, а не к движениям отдельных молекул. Напр., для р-ции нуклеоф. замещения в качестве х можно выбрать антисимметричное колебание в ПС, а в качестве q - коллективную моду, зависящую от инерционной поляризации. В случае р-ций с переносом заряда подходящей оценкой для тq является время продольной релаксации тL; для типичных р-рителей при комнатной т-ре оно равно 10-11-10-12 с. Величину тx обычно оценивают как 10-12 с.
Динамика реагирующей системы и определяемая ею кинетика р-ции задаются системой стохастич. ур-ний для х и q. При тq >> тx устанавливается неравновесный кинетич. режим, при к-ром температурная зависимость константы скорости не является аррениусовской. При тx!тq кинетика р-ции практически равновесна, а константа скорости имеет обычный вид с аррениусовским температурным множителем.
Принципиально интересны системы, для к-рых отнощение тq / тx очень велико; такой кинетич. режим экспериментально обнаружен для р-ций в чрезвычайно вязких средах. Его нельзя описать с помощью константы скорости, т. к. температурная зависимость не описывается одной экспонентой. Описание таких реакций в растворах возможно на основе стохастич. ур-ний с неск. переменными.
Лит.: Энтелис С. Г., Тигер Р. П., Кинетика реакций в жидкой фазе, М., 1973; Мелвин-Хьюз Е. А., Равновесие и кинетика.реакций в растворах, пер. с англ., М., 1975; Эткинс П., в кн.: Физическая химия, пер. с англ., т. 2, М., 1980, с. 439-78; Базилевский М. В., Фаустов В. И., "Успехи химии", 1992, т. 61, в. 7, с. 1185; Chandler D., Introduction to modern statistical mechanics, Oxf., 1987. С.Г. Энтелис, М.В. Базилевский.