Растворы электролитов
РАСТВОРЫ ЭЛЕКТРОЛИТОВ, содержат в заметных концентрациях ионы-катионы и анионы, образующиеся в результате электролитической диссоциации молекул растворенного в-ва. Р-ритель (чистый или смешанный) обычно в сколько-нибудь значит. степени не диссоциирован. Растворы электролитов обладают способностью проводить электрич. ток и относятся к проводникам второго рода. Благодаря увеличению общего числа частиц коллигативные св-ва бесконечно разбавленных растворов электролитов (т. е. св-ва, зависящие только от концентрации растворенного в-ва, но не от его природы) существенно отличаются от тех же св-в растворов неэлектролитов. Этим, в частности, объясняется увеличение осмотич. давления в сравнении со значением, предсказываемым законом Вант-Гоффа (см. Осмос), понижение давления пара р-рителя над р-ром в сравнении с предсказываемым Рауля законом и др. Наличием ионов обусловлены также классификация растворов электролитов, особенности теоретич. подходов в сравнении с др. классами р-ров. Наиб. изучены водные растворы электролитов, играющие важную роль во многих биол., геол. и техн. процессах. Неводные растворы электролитов служат средой для проведения синтеза и электрохим. процессов, используются в совр. технологиях (создание новых химических источников тока, солнечных батарей, процессы разделения в-в и др.).
Классификация растворов электролитов основана на классификации электролитов. Соответственно о растворах электролитов говорят как о симметричных и несимметричных в зависимости от того, распадается ли молекула растворенного в-ва в р-ре на два иона или на большее число частиц; z, z-зарядных [напр., р-р NaCl 1,1-зарядный, р-р СаС12 2,1-зарядный] и т.п.-По степени диссоциации электролита a, к-рая равна отношению числа молекул, диссоциированных на ионы, к полному числу молекул в р-рс, различают сильные (a = 1), слабые (a << 1) электролиты и, соотв., р-ры сильных и слабых электролитов. Такое деление, однако, является условным и отражает состояние электролита в р-ре, определяемое не только природой растворенного в-ва и р-рителя, но и концентрацией (молярной долей хэл), т-рой Т, давлением р.
В зависимости от состояния растворенного в-ва до растворения выделяют два класса растворов электролитов-р-ры ионофоров и р-ры ионогенов. Ионофоры в чистом состоянии существуют в виде ионных кристаллов (напр., галогениды щелочных металлов). В сильнополярных р-рителях (напр., в воде) ионофоры, как правило, диссоциируют полностью и составляют р-ры сильных электролитов. В слабополярных р-рителях (напр., в уксусной к-те) они образуют р-ры слабых электролитов. Ионогены до растворения состоят из молекул, они являются потенц. электролитами, электролитич. диссоциация проходит, как правило, в две стадии и обычно не полностью (хлорная к-та в уксусной к-те).
Растворители для растворов электролитов-как правило, полярные жидкости (чистые или смешанные). Чем больше диэлектрич. проницаемость e р-рителя, тем значительнее ослабляется сильное электростатич. притяжение противоположно заряженных ионов, что способствует возникновению в р-ре ионов. Интенсивное взаимод. последних с молекулами р-рителя приводит к связыванию ионов с молекулами р-рителя (см. Сольватация). Важна также способность молекул р-рителя выступать в качестве доноров или акцепторов протонов или электронов. В зависимости от этих двух св-в различают четыре группы р-рителей: 1) протонные р-рители (вода, спирты, карбоновыс к-ты и др.), к-рые являются хорошими донорами протона и обладают высокой диэлектрич. проницаемостью (e > 15); 2) апротонные дшюлярные р-рители (нек-рые апротонные амиды, кстоны, сульфоксиды и др.), обладающие высокой диэлектрич. проницаемостью, но не обладающие донорно-акцепторными св-вами в отношении протона; 3) электронодонорные-р-рители (напр., эфиры); 4) неполяр-ныс р-рители (сероуглерод, углеводороды), к-рые обладают низкой диэлектрич. проницаемостью (e < 15) и не обладают донорно-акцепторными св-вами ни по отношению к протону, ни по отношению к электрону.
Находящиеся в растворах электролитов ионы могут существовать в виде своб. сольватир. ионов или в виде ассоциатов-контактных или сольватно разделенных ионных пар, тройников и т.д. Поскольку ионные пары не проводят электрич. ток, содержание своб. ионов в растворах электролитов определяется по его электрич. проводимости, в то время как общее число ионов (свободных и ассоциированных) можно определить, напр., спектрофотомет-рич. методами.
Термодинамика растворов электролитов. Равновесные термодинамич. св-ва растворов электролитов описываются парциальными молярными величинами, в к-рых различают катионные и анионные вклады. Напр., для электролита типа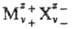 , состоящего из z+-валентных катионов Mz+ и z_-валентных анионов X-, хим. потенциал равен:
, состоящего из z+-валентных катионов Mz+ и z_-валентных анионов X-, хим. потенциал равен:
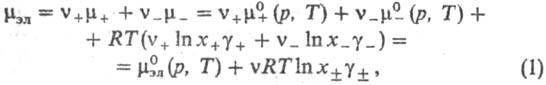
где v=v++v_, x+ и х--молярные доли катионов и анионов соотв., средняя молярная доля электролита хb = =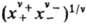 , g+ и g_ -коэффициенты активности катионов и анионов,
, g+ и g_ -коэффициенты активности катионов и анионов, 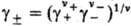 -средний коэф. активности электролита (см. Активность),
-средний коэф. активности электролита (см. Активность),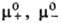 и
и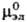 -стандартные хим. потенциалы катионов, анионов и электролита соотв., R-газовая постоянная.
-стандартные хим. потенциалы катионов, анионов и электролита соотв., R-газовая постоянная.
Др. парциальные молярные величины связаны с mэл термо-динамич. соотношениями: парциальная молярная энтропия Sэл = -(9mэл/9Т)p, парциальная молярная энтальпия Hэд_= = -Т2[(9mэл/Т)/9Т]р, теплоемкость Ср = -Т(92mэл/9Т2)p, парциальный молярный объем Vэл = (9mэл/9р)T.
В качестве стандартного состояния компонентов в растворах электролитов выбирают: для р-рителя-состояние чистой жидкости, для растворенного в-ва-состояние в гипотетич. р-ре, где его концентрация и активность равны единице, а термодинамич. св-ва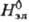 ,
,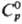 ,
, 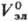 равны соответствующим значениям для бесконечно разб. р-ра.
равны соответствующим значениям для бесконечно разб. р-ра.
В электролитах с неполной степенью диссоциации (а < 1) выражение (1) заменяется соотношением:
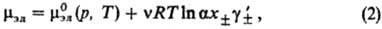
где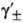 -средний ионный коэф. активности. Степень диссоциации а находят из условия хим. равновесия, к-рое в частном случае симметричного электролита (v+ =v_ = 1) приводит к ур-нию для константы диссоциации KD (или константы ассоциации Ка):
-средний ионный коэф. активности. Степень диссоциации а находят из условия хим. равновесия, к-рое в частном случае симметричного электролита (v+ =v_ = 1) приводит к ур-нию для константы диссоциации KD (или константы ассоциации Ка):
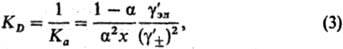
где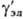 - коэф. активности недиссоциир. молекул (для разбавленных растворов электролитов
- коэф. активности недиссоциир. молекул (для разбавленных растворов электролитов 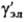 = 1). Аналогично (1) вводится также коэф. активности для р-рителя gS, к-рый связан с ионными коэф. активности Гиббса- Дюгема уравнением.
= 1). Аналогично (1) вводится также коэф. активности для р-рителя gS, к-рый связан с ионными коэф. активности Гиббса- Дюгема уравнением.
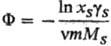
Для описания отклонения от идеального состояния разбавленных растворов электролитов используют кажущиеся осмотические коэффициенты, характеризующие отклонение осмотич. давления от значения, определяемого законом Вант-Гоффа, и связанные с ионными коэф. активности соотношением:
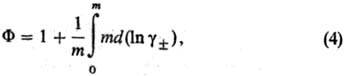
где m - моляльность электролита, gb-ионные коэф. активности в моляльной шкале, MS-молярная масса (кг·моль-1), xS-молярная доля р-рителя.
Хим. потенциалы mэл, коэф. активности gb и осмотич. коэффициенты Ф м. б. определены экспериментально прямыми или косвенными методами: по давлению пара растворенного в-ва или р-рителя, по р-римости, по измерениям эдс электролитич. цепи. Из калориметрич. экспериментов находят парциальную молярную энтальпию Hэл, а из измерений плотности-парциальные молярные объемы Vэл. Поскольку измеримы только суммарные термодинамич. характеристики электролита, для катионов и анионов хим. потенциалы m+ и m_, их стандартные значения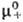 и
и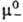 , коэф. активности g+ и g_ и связанные с ними парциальные молярные величины м. б. определены только приближенно, на основе нетер-модинамич. допущений (напр., о равенстве вкладов одинаковых по размерам и степеням окислений катионов и анионов), путем экстраполяции эксперим. данных для различных растворов электролитов с общим катионом или анионом и др.
, коэф. активности g+ и g_ и связанные с ними парциальные молярные величины м. б. определены только приближенно, на основе нетер-модинамич. допущений (напр., о равенстве вкладов одинаковых по размерам и степеням окислений катионов и анионов), путем экстраполяции эксперим. данных для различных растворов электролитов с общим катионом или анионом и др.
Наряду с хим. потенциалами ионов используют также электрохим. потенциал i-го иона с валентностью zi:

где F-Фарадея постоянная, f-внутр. электрич. потенциал раствора электролита (см. Межфазные скачки потенциала).
Статистические теории растворов электролитов основаны на методах стати-стич. механики, их осн. задача-расчет св-в исходя из энергии межчастичного взаимодействия. Развиваются след. подходы: ионный подход (уровень Макмиллана-Майера); ион-но-молекулярный подход (уровень Борна-Оппенгеймера); электрон-ядерный подход (уровень Шрёдингера). Ионный подход является традиционным и к настоящему времени наиб. развит. Он основан на рассмотрении в явном виде только ионов, р-ритель из явного рассмотрения исключается, что требует усреднения ф-ции распределения Гиббса по всем мол. конфигурациям р-рителя (см. Статистическая термодинамика). Энергия межионного взаимод. представляется как сумма слагаемых унарного, бинарного, тернарного и т.д. типов. Унарные слагаемые выражаются через своб. энергию сольватации Wi i-го иона, характеризуемую изменением энергии Гиббса системы при переносе иона из идеальной газовой фазы в бесконечно разб. р-р. Значение Wi совпадает с неидеальной частью стандартного хим. потенциала i-го иона, причем выделяют электростатич., неэлект-ростатич. и хим. вклады в значение Wi. Электростатич. вклад Wiэл м. б. рассчитан согласно модели Борна, в рамках к-рой р-ритель рассматривается как бесструктурная среда, характеризуемая диэлектрич. проницаемостью e:
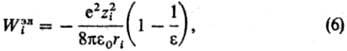
где e0-диэлектрич. постоянная (диэлектрич. проницаемость вакуума), ri-ионный радиус (см. Атомные радиусы). Для уточнения расчета вместо радиуса ri используется эффективный радиус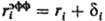 , где через di учитываются размеры молекул р-рителя. Предпринимаются попытки моделирования р-рителя путем введения вместо e диэлектрич. ф-ции и учета нелинейных диэлектрич. эффектов.
, где через di учитываются размеры молекул р-рителя. Предпринимаются попытки моделирования р-рителя путем введения вместо e диэлектрич. ф-ции и учета нелинейных диэлектрич. эффектов.
К неэлектростатич. вкладам в Wi относят индукционный и дисперсионный вклады (см. Дисперсионное взаимодействие), а также вклад, связанный с работой, к-рую необходимо затратить для образования в р-рителе полости и внедрения в нее иона. Расчет этих вкладов производится теми же методами, что и для р-ров неэлектролитов. Для расчета энергии сольватации применяют квантовохим. методы.
Бинарные слагаемые в энергии межионного взаимод. выражаются через потенциалы Wij(R), описывающие эффективное взаимод. ионов сортов i и j, находящихся на расстоянии R друг от друга. Соотв. тернарные слагаемые выражаются через потенциалы, описывающие трехчастичное взаимод. ионов и т. д. Учет бинарных и высших слагаемых в выражении для энергии межионного взаимод. позволяет описывать концентрац. зависимости термодинамич. св-в растворов электролитов.
Св-ва растворов электролитов характеризуются сложными концентрац. зависимостями, обусловленными конкуренцией вкладов разл. типов межчастичных взаимодействий. Обычно ограничиваются учетом потенциалов парного взаимодействия. Его существ. особенность-кулоновский характер межионного взаимод. на больших расстояниях (при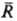 : ,):
: ,):
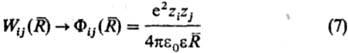
С кулоновским взаимод. связано экранирование межионных взаимод. и образование ионных комплексов, эти процессы характеризуются соотв. радиусом Дебая rD =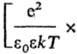
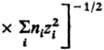 и радиусом Бьеррума
и радиусом Бьеррума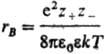 (k-пoстоянная Больцмана, ni-плотность ионов i-го сорта). Первый из них (rD) описывает расстояния, на к-рых экранируется поле иона за счет образования облака ионов противоположного знака, второй (rB)-расстояния, на к-рых кулоновское притяжение между катионом и анионом превышает среднюю энергию теплового движения, что приводит к образованию ионных пар. Эффекты экранирования кулоновских взаимод. учитываются Дебая - Хюккеля теорией; в этой теории первое приближение определяет ионные коэф. активности в области предельных разбавлений (ni : 0). Согласно теории Дебая - Хюккеля, коэф. активности ионов уменьшаются с концентрацией раствора электролита. Наличие в растворах электролитов ионных комплексов учитывается на основе представления о хим. равновесии между свободными и ассоциированными ионами, что приводит к ур-нию, аналогичному (3).
(k-пoстоянная Больцмана, ni-плотность ионов i-го сорта). Первый из них (rD) описывает расстояния, на к-рых экранируется поле иона за счет образования облака ионов противоположного знака, второй (rB)-расстояния, на к-рых кулоновское притяжение между катионом и анионом превышает среднюю энергию теплового движения, что приводит к образованию ионных пар. Эффекты экранирования кулоновских взаимод. учитываются Дебая - Хюккеля теорией; в этой теории первое приближение определяет ионные коэф. активности в области предельных разбавлений (ni : 0). Согласно теории Дебая - Хюккеля, коэф. активности ионов уменьшаются с концентрацией раствора электролита. Наличие в растворах электролитов ионных комплексов учитывается на основе представления о хим. равновесии между свободными и ассоциированными ионами, что приводит к ур-нию, аналогичному (3).
С увеличением концентрации электролита возникает необходимость учитывать и некулоновскую часть межионного взаимод., для чего прибегают к нек-рым моделям. При этом наряду с индукционным, дисперсионным, обменным и др. видами межчастичных взаимод. некулоновский потенциал учитывает сольватац. эффекты, связанные с влиянием р-ри-теля. В частности, учет некулоновской части взаимод. стабилизирует уменьшение коэф. активности ионов с концентрацией и может объяснить их увеличение, наблюдаемое экспериментально. Наипростейшей ионной моделью растворы электролитов является модель заряженных твердых сфер (т. наз. примитивная модель). Первые попытки описания примитивной модели были выполнены в рамках теории Дебая-Хюккеля (второе приближение). Более корректно учет размера ионов и неку-лоновского взаимод. осуществляется на основе методов статистич. термодинамики (см. Жидкость).
В рамках примитивной модели размеры ионов отличаются от кристаллографич. радиусов из-за сольватац. эффектов. Однако даже при одном и том же выборе размеров ионов удовлетворительно описать эксперим. результаты для разл. термодинамич. св-в растворах электролитов в примитивной модели оказалось затруднительным. К более корректным результатам приводит модель парного взаимод. типа "прямоугольной ямы", в к-рой ширина потенц. ямы выбирается равной диаметру молекулы р-рителя, а глубина ямы считается подгоночным параметром, учитывающим сольватац. эффекты; при этом используются кристаллографич. размеры ионов. В более реалистич. модели Фридмана в некулонов-ском потенциале межионного взаимод. выделяют три слагаемых, соответствующих: 1) главному отталкиванию ионов, определяемому их кристаллографич. размерами; 2) эффекту поляризации полости ионов р-рителем; 3) потенциалу Гер-ни-Франка, описывающему структурные эффекты, связанные с перекрыванием сольватных оболочек ионов при их сближении. Расчеты на основе ион-молекулярных моделей показывают, что на малых расстояниях межионные потенциалы имеют отталкивательный характер, на больших расстояниях, в соответствии с (7), зависят от расстояния между ионами асимптотически, как и при кулоновском взаимод., на промежут. расстояниях осциллируют вблизи этой асимптоты, причем с уменьшением размера иона (или с увеличением его валентности) амплитуда осцилляции возрастает, что соответствует усилению роли сольватац. эффектов.
Пренебрежение трехчастичными (и высшими) межионными взаимод. ограничивает возможности ионного подхода. В частности, для 1,1-зарядных водных растворов электролитов ионный подход обеспечивает количеств. описание термодинамич. св-в в области концентраций до 1 М. Учет концентрац. зависимости диэлектрич. проницаемости позволяет немного расширить эту концентрац. область. Формально расширения области применимости ионного подхода можно достигнуть, дополняя полученные с его помощью результаты разл. эмпирич. поправками. Примером такого подхода может служить полуэмпирич. ур-ние Питцера для осмотич. коэффициента или метод Робинсона-Стокса описания ионных коэф. активности с учетом гидратации (с помощью гидра-тац. чисел). Для описания многокомпонентных растворов электролитов широко используется правило Здановского, основанное на предположении о том, что смешение изописстич. р-ров разл. электролитов, химически не взаимодействующих между собой, происходит без изменения активности р-рителя.
Ионно-молекулярный подход основан на рассмотрении в явном виде как ионов, так и молекул р-рителя. Главные результаты получены в 70-80-х гг. 20 в. на базе расчетных методов, интенсивно развиваемых в теории жидкостей. Это в осн. метод интегральных ур-ний для корреляц. ф-ций, метод кластерных разложений, теория возмущений, а также компьютерное моделирование. Благодаря явному учету ионно-молекулярных и межмолекулярных взаимод. возможно описание не только термодинамич., но и структурных св-в растворов электролитов. В частности, важный результат - описание сольватации ионов в зависимости от концентрации и др. параметров р-ра, объяснение концентрационных, температурных и барич. зависимостей св-в в широких интервалах состава, т-ры и давления.
Наипростейшей ион-молекулярной моделью растворов электролитов является ион-дипольная модель, в к-рой ионы рассматриваются как заряженные твердые сферы, а молекулы р-рителя моделируются твердыми сферами с дипольным моментом. Полученные выражения для термодинамич. ф-ций обобщают ур-ния, используемые в ионном подходе. В частности, в предельном случае малых концентраций выражения для ионных коэф. активности включают члены, основанные на теории Дебая-Хюккеля, а выражения для энергии сольватации-борновскую ф-лу (6) с эффективным радиусом иона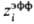 , в к-ром поправка di в явном виде зависит от диэлектрич. проницаемости р-рителя и соотношения размеров иона и молекулы. Выражение для диэлектрич. проницаемости удовлетворительно описывает эффект ее уменьшения при увеличении концентрации ионов.
, в к-ром поправка di в явном виде зависит от диэлектрич. проницаемости р-рителя и соотношения размеров иона и молекулы. Выражение для диэлектрич. проницаемости удовлетворительно описывает эффект ее уменьшения при увеличении концентрации ионов.
Предпринимаются попытки учета квадрупольного элект-рич. момента и поляризуемости молекул р-рителя, а также взаимод., ответственных за образование в растворах электролитов ассоциатов и сольватов. Наиб. реальные модели разработаны в осн. для водных растворов электролитов и базируются обычно на компьютерном моделировании. Для описания ионно-молекулярных и межмолекулярных взаимод. применяют эмпирич. модели воды (модель ST2, модель центр. сил и др.), а также модели, основанные на квантовохим. расчетах. Рассчитанные парциальные радиальные ф-ции распределения дают информацию о структуре р-ра. В частности, с помощью ионно-молекулярных ф-ций определяют координац. числа сольватации. Найденные с помощью парциальных радиальных ф-ций структурные факторы удовлетворительно согласуются с данными дифракц. измерений.
Электрон-ядерный подход основан на учете электроста-тич. взаимод. между электронами и ядрами, входящими в состав ионов и молекул в растворах электролитов. Этот подход является наиб. последовательным, он основан на квантовомех. рассмотрении и разработан пока лишь для ион-молекулярных комплексов.
Важное значение в физ. химии растворов электролитов имеют исследования транспортных св-в, особенно электрич. проводимости (см. Электропроводность электролитов). Наличие ионов заметно сказывается на диффузии, вязкости, теплопроводности.
Лит.: Робинсон Р. А., Стоке Р. Г., Растворы электролитов, пер. с англ., М., 1963; Измайлов Н.А., Электрохимия растворов, 3 изд., М., 1976; Мищенко К. П., Полторацкий Г.М., Термодинамика и строение водных и неводных растворов электролитов, 2 изд., Л., 1976; Юхновский И. Р., Головко М.Ф., Статистическая теория классических равновесных систем, К., 1980; Ионная сольватация, М., 1987; Falkenhagen H., Theorie der Elektrolyte, Lpz., 1971; The chemical physics of solvation, pt А, В, С, Amst., 1985-88.
М. Ф. Головко.