Отличительные свойства органических соединений и особенности органических реакций
Многообразие органических соединений обусловлено специфическими особенностями углерода сравнительно с другими химическими элементами:
1. Углерод способен соединяться с большинством других элементов. Это свойство углерода, связанное с его положением в периодической системе элементов Д. И. Менделеева, обусловлено его почти электронейтральным характером и способностью образовывать ковалентные связи.
2. Атомы углерода способны также соединяться друг с другом, образуя углеродные цепи различного вида (прямые, разветвленные, замкнутые в цикл). Таким же свойством обладают некоторые другие элементы, однако все они образуют цепи лишь с небольшим числом звеньев. Так, для кислорода известны цепи максимально из трех атомов, для азота — из четырех, для кремния — из шести атомов. Для углерода, в отличие от всех остальных элементов, способность атомов соединяться друг с другом, по-видимому, совершенно не ограничена. В настоящее время получены высокомолекулярные продукты углеводородного характера, углеродная цепь которых состоит из сотен и тысяч углеродных атомов (полиэтилен, полиизобутилен и др.).
3. На определенной стадии усложнения органических веществ появляется возможность изомерии; вследствие этого одному и тому же составу соответствует не одно, а несколько веществ, отличающихся друг от друга только химическим строением.
Хотя число органических соединений чрезвычайно велико иони весьма разнообразны, можно выделить ряд общих характерных свойств, отличающих органические соединения от неорганических.
Почти все органические соединения, за исключением сравнительно немногих (например, четыреххлористого углерода, фторуглеродов), горючи. Большинство неорганических соединений не горит.
Органические соединения обычно представляют собой газы, жидкости или низкоплавкие твердые вещества. Огромное большинство твердых органических веществ плавится в интервале сравнительно невысоких температур (от комнатной до 400 °С). Большая часть неорганических соединений представляет собой твердые вещества, плавящиеся при весьма высоких температурах.
В качестве примеров типично неорганического и типично органического вещества можно взять хлористый натрий и хлористый этил, молекулярные веса которых примерно одинаковы (58,44 и 64,51):
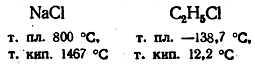
Такие резкие отличия в физических свойствах типично органических и типично неорганических соединений обусловлены отличием в их строении, а именно: неорганические соединения большей частью построены ионно, а органические соединения — ковалентно.
Особенности строения органических соединений как соединений с ковалентными связями находят свое отражение не только в физических, но и в химических свойствах. Так, реакции неорганических соединений (электролитов) в водных растворах, например
NaCl + AgNO3 -----> AgCl + NaNO3
протекают с большой скоростью, практически мгновенно, в то время как реакции органических соединений, например
С2Н5Cl + NaOH ------> NaCl + С2Н5ОН
идут значительно медленнее. Другой отличительной особенностью органических реакций является то, что они редко протекают количественно. Если реакции нейтрализации, осаждения, окисления и восстановления неорганических веществ протекают с количественным выходом, то при органических реакциях очень хорошим считается выход 85—90% от теоретического.
Типично неорганические соединения реагируют в виде ионов, которые образуются вследствие электролитической диссоциации. Это обусловливает большую скорость неорганических реакций, так как время реакции определяется только числом столкновений противоположно заряженных ионов в единицу времени, а это число составляет примерно 1011 в секунду.
Для того чтобы понять причины медленного протекания реакций ковалентно построенных органических соединений, необходимо выяснить механизм этих реакций.
Течение химических реакций в общем случае определяется двумя факторами: изменением свободной энергии системы и скоростью превращения. Первый фактор — термодинамический, второй — кинетический.
Для практически необратимых реакций решающее значение имеет скорость превращения; поэтому термодинамически менее выгодное, но все же возможное направление часто доминирует вследствие его большей скорости. При достаточно быстро протекающих обратимых реакциях главным фактором является термодинамический, так как положение равновесия не зависит от скорости его установления, а определяется лишь отношением констант скорости прямого и обратного процесса. В случае медленно протекающих обратимых реакций играют роль оба фактора.
Органические реакции лишь в сравнительно редких случаях протекают до стадии установления равновесия; вследствие этого результаты сложных процессов чаще определяются кинетикой составляющих реакций, чем термодинамикой. Однако в общем случае заметное влияние оказывают оба фактора, что в большой мере затрудняет изучение и трактовку течения химических реакций.
Согласно основному уравнению кинетики химических реакций
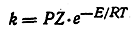
константа скорости k является функцией температуры T, числа столкновений Z между реагирующими молекулами, вероятностного фактора Р и энергии активации Е (R — газовая постоянная, е — основание натуральных логарифмов). Термохимические исследования показывают, что энергии активации реакций органических соединений в большинстве случаев меньше энергий их диссоциации на ионы. Таким образом, органические реакции обычно протекают не через стадию образования ионов, а иными путями.
Представление о причинах характерных особенностей органических реакций дает теория переходного состояния, или активного комплекса.
Теория переходного состояния может быть уяснена, например, из рассмотрения бимолекулярной реакции замещения:
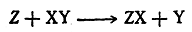
При взаимодействии реагента Z с молекулой XY возможны самые различные столкновения этих двух частиц. Однако в простейшем случае химическая реакция происходит легче всего, если Z приближается к молекуле XY вдоль ее оси (или под небольшим углом к этой оси) с той стороны, где расположен X:
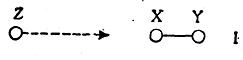
В определенный момент сближения этих двух частиц между Z и X образуется слабая связь при одновременном ослаблении связи X—Y:
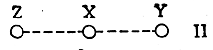
В этом состоянии X уже в известной мере связан с Z и в то же время еще не потерял связи с Y. Такое состояние и носит название переходного состояния, или активного комплекса.
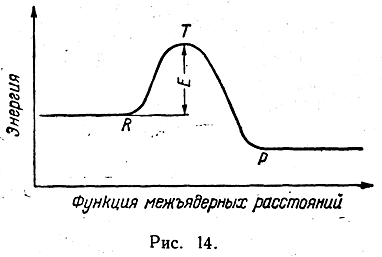
Так как для достижения такого состояния при сближении частиц Z и XY необходима затрата энергии, в частности, на растягивание связи X—Y и на преодоление отталкивания атома Z молекулой XY, то переходное состояние соответствует максимуму на кривой, изображающей энергетическое состояние реагирующей системы (рис. 14). При дальнейшем сближении Z и X между ними образуется ковалентная связь, а связь X с Y окончательно разрывается:
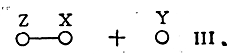
Этот процесс сопровождается понижением энергии реагирующей системы. Точка R на кривой (рис. 14) соответствует энергии системы, когда Z и XY удалены друг от друга (I), точка Р — аналогичной системе ZX и Y (III), точка Т — энергии переходного состояния (II). Разность между энергией переходного состояния Т и энергией первоначальной системы R составляет энергию активации Е данной реакции. Таким образом, энергия активации представляет собой энергию, необходимую для приведения двух реагирующих частиц в состояние активного комплекса.
С точки зрения изложенных представлений, органическая реакция, например действие щелочи на хлористый этил, протекает следующим образом:
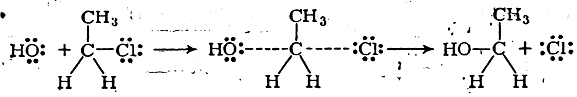
Следует отметить, что переходное состояние является принципиально неустойчивой системой. Оно соответствует максимуму на энергетической кривой, и поэтому такую систему нельзя рассматривать как какое-то промежуточное соединение. Всякое промежуточное химическое соединение, пусть малоустойчивое, соответствует относительному минимуму энергии, для перехода из которого в конечное состояние необходимо вновь преодолеть некоторый энергетический барьер (второй максимум — второе переходное состояние). Промежуточные соединения являются настоящими химическими соединениями, переходное же состояние не является химическим соединением в обычном смысле слова, поэтому к нему неприменимы такие понятия и закономерности, связанные с настоящими химическими соединениями, как валентность, постоянство атомных расстояний и углов валентностей и т. д.
За исключением ряда специальных случаев, первостепенное значение в органических реакциях играет энергия активации Е. Входя в показатель степени основного кинетического уравнения, Е самым существенным образом определяет скорость реакции, причем сравнительно небольшие изменения Е изменяют скорость реакции в десятки раз.
Необходимо, однако, указать, что расчетный аппарат теории переходного состояния в настоящее время находится на низком уровне, и в большинстве случаев невозможно вычисление энергии активации с такой степенью точности, чтобы можно было объяснить (не говоря уже о предсказании) преобладающее протекание одних органических реакций сравнительно с другими возможными. В связи с этим в настоящее время единственным способом составить суждение о факторах, определяющих энергию активации, например конкурирующих органических реакций, являются чисто качественные сопоставления строения активных комплексов конкурирующих реакций и сравнительная оценка их энергии.
В развитие основных положений теории химического строения о связи реакционной способности органических веществ с их химическим строением в органической химии были созданы представления о гетеролитических и гомолитических реакциях.
Для гетеролитических реакций характерен такой разрыв химических связей у реагирующих веществ, при котором электронная пара, осуществляющая ковалентную связь, целиком остается у одного из двух ранее связанных атомов, например:
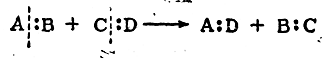
Частным случаем гетеролитических реакций являются ион-ные реакции, протекающие не через активный комплекс, а с образованием свободных ионов как кинетически независимых частиц.
Для гомолитических реакций характерен разрыв химических связей с разделением электронной пары, осуществлявшей ковалентную связь, например:
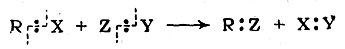
Частным случаем гомолитических реакций являются свобод-норадикальные реакции, протекающие с образованием свободных радикалов как кинетически независимых частиц.
Гетеролитические реакции, в том числе и протекающие через стадию образования свободных органических ионов, весьма многочисленны. К ним, по-видимому, относится большая часть известных в настоящее время органических реакций, например реакции этерификации и гидролиза, протекающие в присутствии кислотных и основных катализаторов, присоединение галоидов к олефинам в полярных растворителях, присоединение синильной кислоты и ряда других реагентов к альдегидам и кетонам, разнообразные прототропные таутомерные превращения и многие другие реакции.
Следует отметить, что огромное большинство органических гетеролитических реакций, в том числе и ионных, протекает значительно медленнее ионных неорганических реакций. Причина этого явления становится понятной, например, при рассмотрении взаимодействия альдегидов с синильной кислотой:
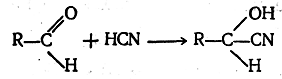
Как установлено, первая стадия этой реакции заключается в присоединении аниона CN- к карбонильному углеродному атому альдегида:
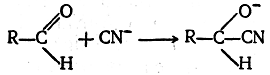
Эта стадия реакции представляет собой процесс взаимодействия не аниона с катионом, а аниона с нейтральной молекулой; следовательно, она будет протекать медленнее тех процессов, в которых участвуют только ионы.
Таким образом, несмотря на то, что вторая стадия циангид-ринной реакции, представляющая собой взаимодействие аниона с протоном
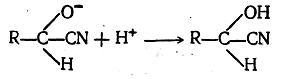
протекает практически мгновенно, в целом присоединение HCN к альдегидам или к кетонам протекает сравнительно медленно. Наличием стадии взаимодействия иона с нейтральной молекулой и объясняется медленность большинства ионных органических реакций сравнительно с ионными неорганическими реакциями.
Открытие гомолитических и, в частности, свободнорадикаль-ных реакций сыграло важную роль в развитии теории химического строения. Оказалось, что многие органические реакции, в частности распад диазосоединений, распад перекисей, реакции галоидирования, нитрования и окисления парафинов и алицик-лических углеводородов, протекают с образованием свободных радикалов. Свободные радикалы, как правило, более активны, чем молекулы с четным числом валентных электронов. Они легко взаимодействуют не только друг с другом (рекомбинация и диспропорционирование), но и главным образом с недиссо-циированными молекулами, образуя при этом новый свободный радикал, который в свою очередь реагирует с молекулой и т. д. Таким образом, свободный радикал, возникнув однажды, вызывает цепь превращений, которая обрывается рекомбинацией радикалов или каким-либо другим способом.
Простым примером цепной органической реакции может служить хлорирование метана, происходящее на свету
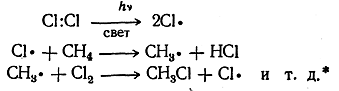
Развитие учения о цепных реакциях является крупным достижением современной химической науки. В настоящее время это учение развивается главным образом школами Н. Н. Семенова в СССР и Хиншельвуда в Англии. Н, Н. Семеновым, в частности, создана теория разветвленных цепных реакций.
Теория цепных реакций способствовала устранению упрощенных представлений о многих органических реакциях как о более или менее легко происходящих непосредственных перегруппировках связей при встрече двух молекул и дала ключ к выяснению механизма большого числа органических реакций.