Радикальные реакции
РАДИКАЛЬНЫЕ РЕАКЦИИ, осуществляются с участием радикального центра своб. радикалов.
Характеризуются, как правило, большими значениями предэкспоненц. множителя в ур-нии Аррениуса и малыми энергиями активации Е. Включают по крайней мере две стадии - образование своб. радикалов и их гибель. Образование своб. радикалов происходит вследствие разрыва хим. связи, при к-ром на обоих фрагментах молекулы или на одном из них остается по одному неспаренному электрону, напр.:
Эта р-ция м. б. индуцирована нагреванием, облучением, действием восстановителей или др. путями.
Радикалы обладают разл. реакц. способностью, зависящей от их природы, типа р-ций, полярного и стерич. факторов, эффекта р-рителя.
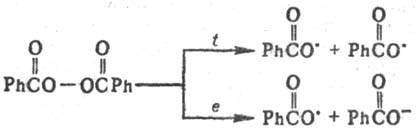
Реакции между свободными радикалами. Осуществляются при взаимод. двух одинаковых или разл. радикалов с образованием новой хим. связи (рекомбинация). Р-ция обычно диффузионно контролируемая; Е от 0 до 5 кДж/моль. Происходит, напр., при дегидродимеризации орг. соедине-ний под действием пероксидов или др. окислителей, напр.:
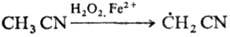
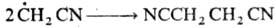
Др. пример рекомбинации своб. радикалов - электрохим. анодный синтез углеводородов (см. Кольбе реакции):
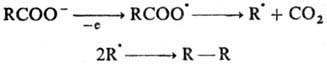
Рекомбинация в -катодном процессе может происходить при электрохйм. восстановлении, напр., карбонильных соед.:
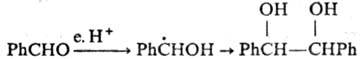
При близком расположении двух своб. радикалов в "клетке" р-рителя (напр., генерируемых при термораспаде пероксидов или диазосоединений) р-ция осуществляется с высокой селективностью (см. Клетки эффект). Влияние этого эффекта уменьшается с увеличением т-ры и понижением вязкости р-рителя.
При взаимод. двух радикалов возможно также диспропор-ционирование- перенос атома Н (реже-атома галогена) от одного радикала к другому, напр.:

Диспропорционирование, как правило, более медленный процесс, чем рекомбинация. Обе р-ции экзотермичны и нередко идут одновременно.
Реакции свободных радикалов с молекулами. Характерны для таких практически важных процессов, как хлорирование, бромирование, сульфохлорированис, автоокисление, полимеризация и др., протекающих по цепному механизму (см. Цепные реакции). Их константы скорости на неск. порядков ниже, чем для рекомбинации; Е = 20-60 кДж/моль. Так, р-ция автоокисления осуществляется в результате многократно повторяющихся р-ций по схеме:
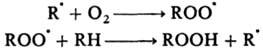
Обрыв цепи происходит путем рекомбинации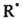 с
с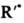 или с ROO•. Для торможения автоокислит. процессов используют ингибиторы, обычно пространственно-затрудненные фенолы или ароматич. амины. Радикалы, взаимодействуя с ингибиторами, образуют неактивные феноксильные или аминильные радикалы, к-рые не способны участвовать в стадиях роста цепи. Механизм, подобный описанному, обусловливает защитные св-ва витамина Е при действии О2 на клетки живых организмов.
или с ROO•. Для торможения автоокислит. процессов используют ингибиторы, обычно пространственно-затрудненные фенолы или ароматич. амины. Радикалы, взаимодействуя с ингибиторами, образуют неактивные феноксильные или аминильные радикалы, к-рые не способны участвовать в стадиях роста цепи. Механизм, подобный описанному, обусловливает защитные св-ва витамина Е при действии О2 на клетки живых организмов.
Механизм р-ции замещения радикалами алкильных, тиильных, ацилоксильных, алкоксильных и нек-рых др. групп включает разрыв связи в исходной молекуле и образование новой связи с участием радикала, напр.:
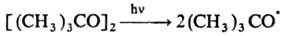
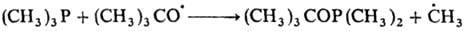
По аналогии с нуклеоф. р-циями замещения такой механизм наз. механизмом SR2.
Стереохимия р-ции зависит от характера субстрата. Обычно образующийся радикал с неспаренным электроном у хирального атома С имеет плоскую или близкую к плоской конфигурацию, что приводит к рацемизации. В циклич. системах инверсия радикального центра затруднена, особенно в случае малых циклов, напр.:
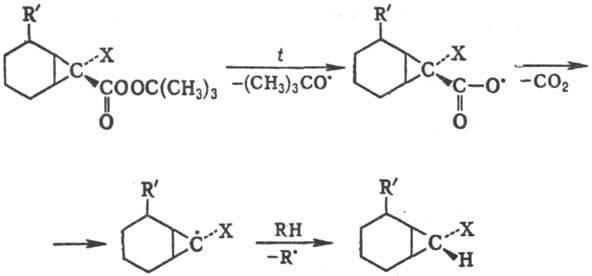
Р-ции радикального замещения в ароматич. ядре обычно не являются цепными и осуществляются через стадию образования циклогексадиенильных радикалов:
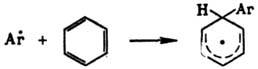
Превращение в конечный продукт происходит в результате отрыва атома Н (р-ция 1); побочные процессы-рекомбинация (2) и диспропорционирование (3):
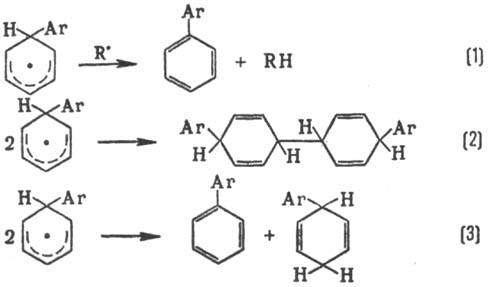
Электронедонорные и электроноакцепторные заместители активируют ароматич. ядро. В р-циях образуются о, м, и n-изомеры с преобладанием о-изомера.
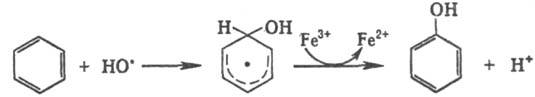
Гидроксильные радикалы, к-рые генерируются реактивом Фентона (Н2О2+Fe2+), способны атаковать неактивир. ароматич. ядро. При этом образуется радикал, к-рый диме-ризуется или в присут. Fe3+ превращ. в фенол:
Среди р-ций радикального замещения в ароматич. ядро практич. значение имеет алкилирование протонир. гетеро-ароматич. соединений с атомом N в кольце. В пиридинах и хинолинах замещение происходит у атомов С в положениях 2 и 4.
Свободнорадикальное присоединение по связи С=С-цепной процесс с короткими кинетич. цепями. Инициируется пероксидами, ионами переходных металлов и УФ облучением. Р-ция нестереоселективна для алкенов, высокостерео-селективна (транс-присоединение) для циклоалкенов; Е обычно не превышает 40 кДж/моль.
К 1-алкенам радикал обычно присоединяется по а-атому С, напр.:
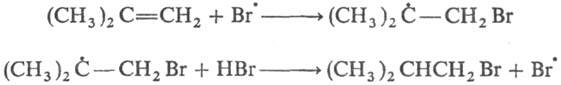
Свободнорадикальное присоединение НВr к несимметричным олефинам идет против правила Марковникова. Это объясняется тем, что в случае атаки такого олефина р-ция осуществляется по пути образования более стабильного радикала (увеличение числа алкильных заместителей при радикальном центре увеличивает стабильность радикала), поболее доступному атому С и па месту наиб. электронной плотности в молекуле, напр.:
Свободнорадикальное присоединение по связи С=С широко используется в органическом синтезе, в т.ч. в промышленности.
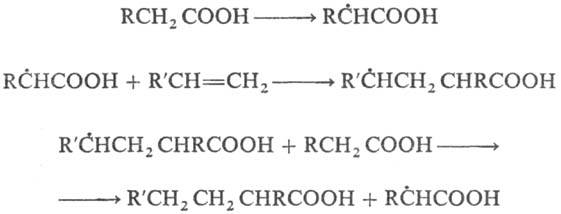
К реакциям свободнорадикального присоединения относится также теломеризация - цепная реакция непредельных соед. в присут. в-в (телогенов), в результате к-рой образуется смесь низкомол. гомологов (теломе-ров). Реакция используется в пром-сти для получения a, a, a, w-тетрахлоралканов из СС14 и этилена и разветвленных карбоновых к-т из пропионовой к-ты и этилена, напр.:
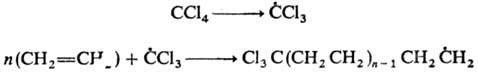
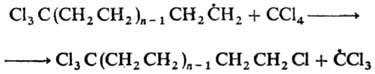
По аналогичному механизму осуществляется радикальная полимеризация непредельных соединений.
Внутримолекулярные реакции свободных радикалов. К этому типу р-ций относится фрагментация - распад радикала на молекулу и радикал с меньшей мол. массой. Р-ция характерна для мн. b-галоген-, b-тио-, a-гидроксиалкильных радикалов, а также для алкоксильных, ацилоксильных и нек-рых др. радикалов. Если фрагментация возможна по неск. направлениям (напр., при окислении несимметричных третичных спиртов), реализуется преим. то, к-рое приводит к более стабильному радикалу, напр.:
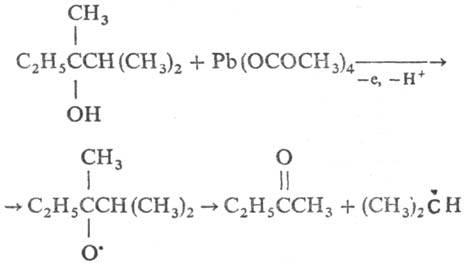
Т.к. в р-циях этого типа гемолизу подвергается связь, обычно расположенная в b-положении по отношению к неспаренному электрону, то такой процесс иногда наз. b-рас-щеплением или b-распадом. Ацильные радикалы, генерируемые из альдегидов, легко фрагментируют при т-ре выше 100°С на алкильные радикалы и СО.
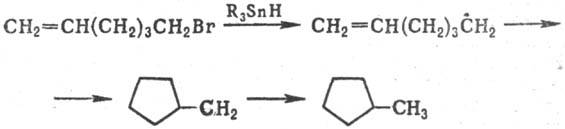
Фрагментацию циклоалкоксильных радикалов, генерируемых из циклоалканолов или циклоалканонов, используют в синтезе алифатич. бифункцион. соединений по схеме:
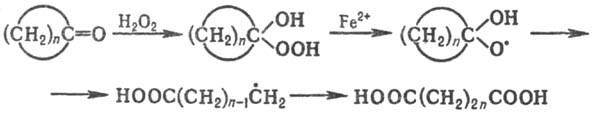
Фрагментация промежуточно образующихся алкильных радикалов происходит при получении этилена путем пиролиза нефтяных углеводородов.
Радикалы могут претерпевать перегруппировки с миграцией атомов и(или) функц. групп. При этом всегда происходит образование более стабильного радикала, чем исходный. Для алкильных, алкоксильных, аминилъных, амидиль-ных радикалов характерна 1,5-миграция атома Н от атома С к радикальному центру. Эта р-ция - ключевая стадия при окислении алканолов в тетрагидрофураны, карбоновых к-т в g-лактоны, метилалкилкетонов в g-дикетоны, напр.:
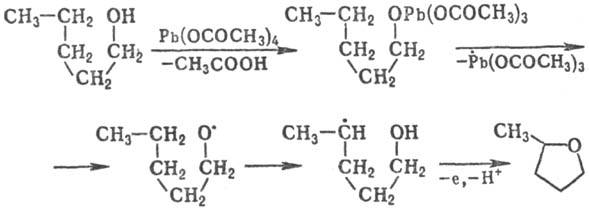
Иногда наблюдается наряду с 1,5- также и 1,6-миграция атомов Н, значительно реже-1,3- и 1,4-миграции.
Известны примеры р-ций С-центрированных радикалов с 1,4-миграцией арилов или группы CN, с 1,2-миграцией атомов С1, Вr или арильных, ацетоксилъных, ацильных и винильных группировок, напр.:
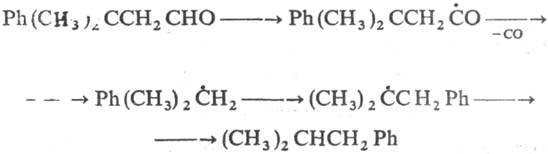
Известно также внутримол. присоединение углерод- или кислород центрированных радикалов к кратным связям или к ароматич. ядру. Таким путем легко образуются 5- и 6-членные циклы, напр.:
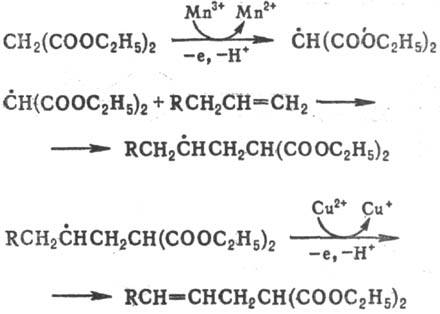
Окисление и восстановление свободных радикалов. Наиб. известно одноэлектронное окисление С-центрированных радикалов солями Сu(II), Co(III), Mn(III), Fe(III), Pb(IV), Се (IV). Р-ция осуществляется с высокими скоростями с переносом лиганда (С1, Вг, CN и др.), напр.:
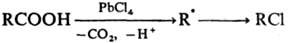
При окислении ацетатом или сульфатом Сu(II) р-ция сопровождается депротонированием, что приводит к образованию двойной связи; р-ция используется в синтезе непредельных соед. с функц. группами, напр.:
Восстановление-типичная р-ция феноксильных и нйтро-ксильных радикалов, напр.:
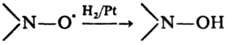
Относительно легко восстанавливаются по механизму переноса электрона С-центрированные радикалы с электро-ноакцепторными заместителями при радикальном центре, напр.:
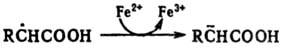
См. также Радикалы свободные.
Лит.: НонхибелД., Уолтон Дж., Химия свободных радикалов, пер. с англ., М., 1977; Розанцев Э.Г., Шолле В. Д., Органическая химия свободных радикалов, М., 1979; Девис Д., Перрет М., Свободные радикалы в органическом синтезе, пер. с англ., М., 1980; Нонхибел Д., Теддер Дж., Уолтон Дж., Радикалы, пер. с англ., М., 1982; Free radicals, ed. by J.K. Kochi, v. 1-4, N.Y., 1973-80; Selectivity and synthetic applications of radical reactions. Tetrahedrpn symposia-in-print, "Tetrahedron", 1985, v. 41, № 19; Giese В., Radical in organic synthesis. Formation of carbon bonds, Oxf., 1986; Curran D.P., "Synthesis", 1988, № 6, p. 417-39; там же, 1988, № 7, p. 489-513.
Г. И. Никишин.